
- Title
-
Transcriptome remodelling and changes in growth and cardiometabolic phenotype result following Grb10a knockdown in the early life of the zebrafish
- Authors
- Evans, B.L., Garner, T., De Leonibus, C., Wright, L., Sharps, M., Wearing, O.H., Ripley, D.M., Shiels, H.A., Hurlstone, A.F.L., Clayton, P.E., Stevens, A.
- Source
- Full text @ Cell. Mol. Life Sci.
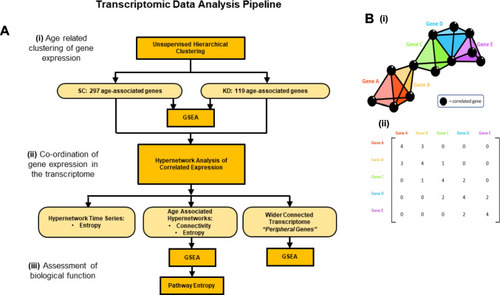
Pipeline of transcriptomic analysis and hypergraphs. ( |
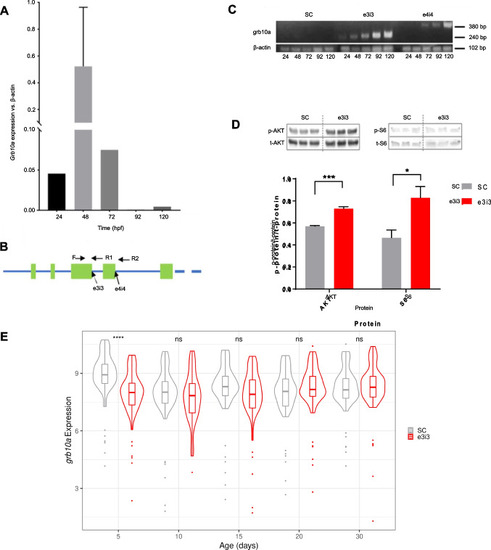
Grb10a is successfully knocked down in zebrafish injected with splice-blocking antisense oligonucleotides. ( |
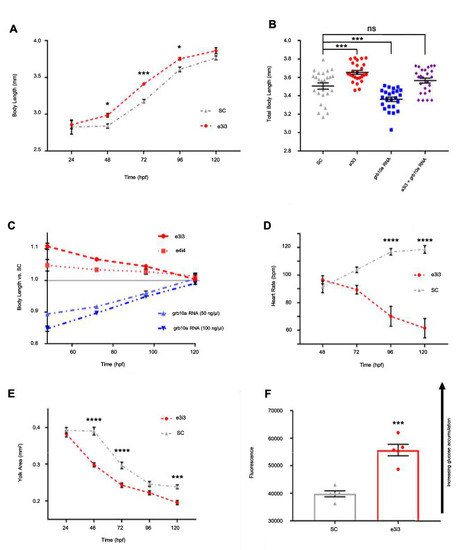
Growth and cardiometabolic phenotype are significantly impacted by early life grb10a knockdown. ( |

Transcriptomic analysis of standard control and grb10a knockdown gene expression over the first 30 dpf. Hierarchically clustered heat maps of gene expression generated from an Affymetrix GeneChip™ Zebrafish Genome Array of SC and KD zebrafish RNA, taken at 5, 10, 15, 20, and 30 dpf. ( |
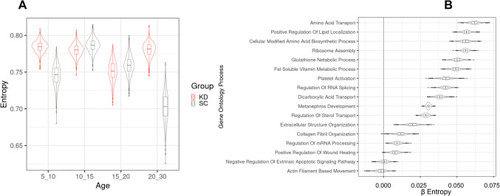
Hypergraph entropy in the transcriptome reveals differences over time and between functions. ( |
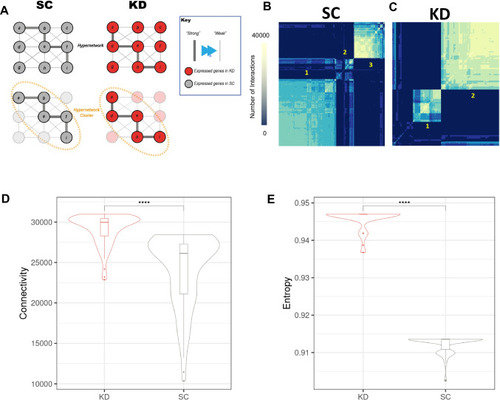
The coordination of age associated genes is altered by grb10a KD, as assessed by hypergraph analysis ( |
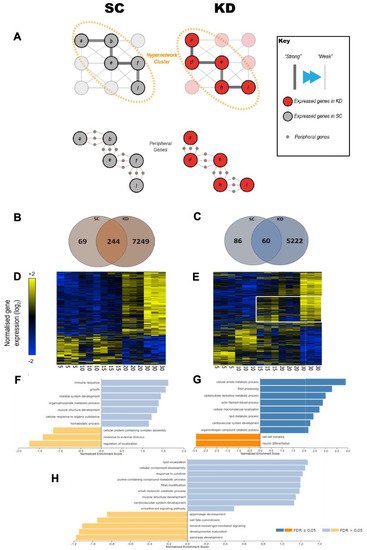
Analysis of the set of genes in the wider transcriptome shows a 27.8-fold increase in the KD ZF. ( |
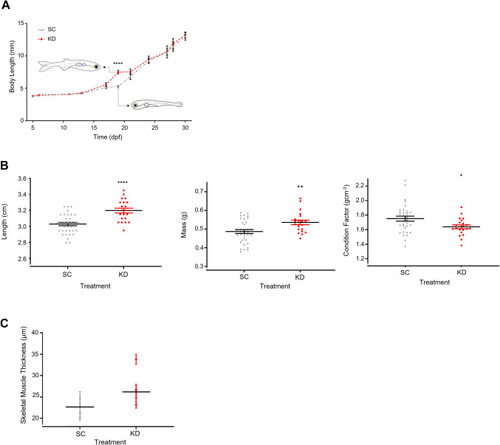
Growth in juvenile Zebrafish and adult size and skeletal muscle thickness at 18 months. ( |
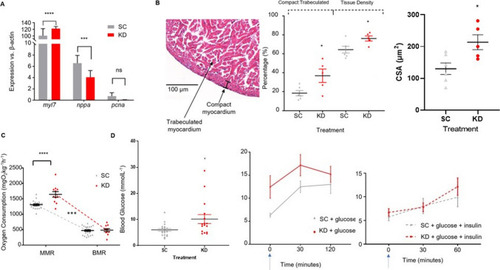
Cardiac gene expression and Cardiometabolic function in 18-month-old adult Zebrafish. ( |
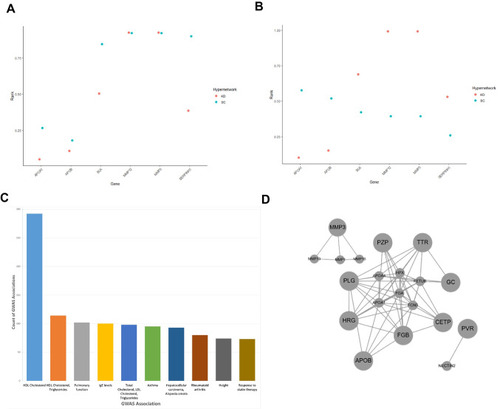
Genes associated with cardio-metabolic diseases. Cardio-metabolic disease GWAS genes ranked by normalized row sum in the age-related transcriptome hypergraphs of control and GRB10 knockdown zebrafish. Six genes out of 29 differentially expressed genes were present in both the SC and KD were shown to be related to cardiometabolic health. |