
- Title
-
Deficiency of heme oxygenase 1a causes detrimental effects on cardiac function
- Authors
- Wang, H., Siren, J., Perttunen, S., Immonen, K., Chen, Y.C., Narumanchi, S., Kosonen, R., Paavola, J., Laine, M., Tikkanen, I., Lakkisto, P.
- Source
- Full text @ J. Cell. Mol. Med.
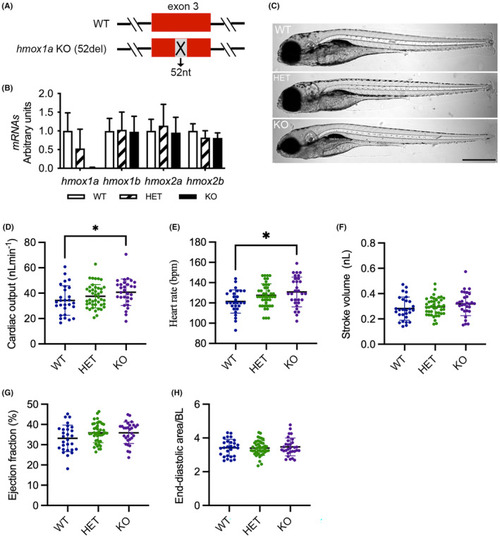
Deletion of |
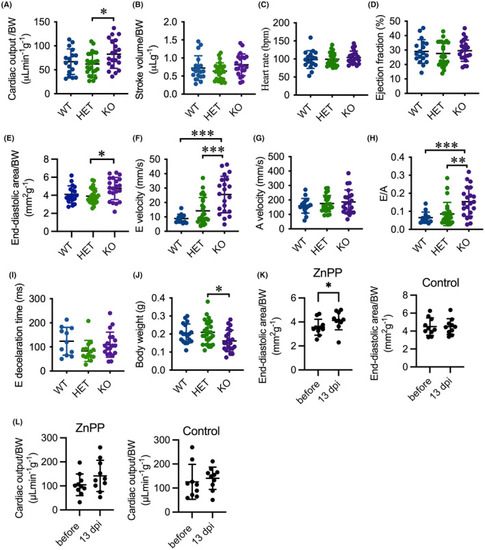
Deletion of |
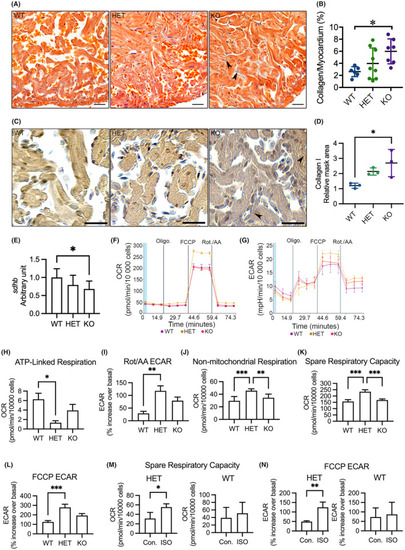
Analyses of myocardial interstitial fibrosis, cardiac OXPHOS gene expression and mitochondrial respiration in adult cardiomyocytes. (A) Representative images of acid fuchsin orange G (AFOG) staining of ventricular sections showing accumulation of collagen (blue) in the myocardium (orange). Arrowhead indicates accumulated collagen. (B) Quantification of AFOG staining indicating increased collagen accumulation in the myocardium in KO(52del). WT |
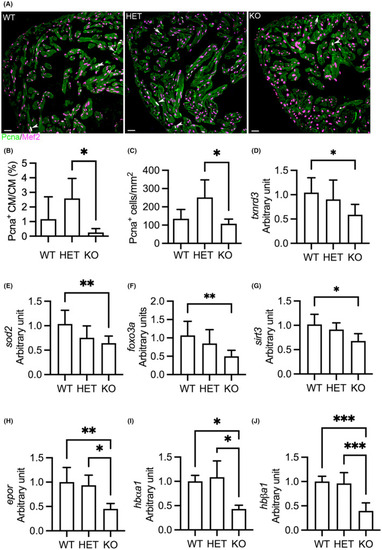
Deletion of |
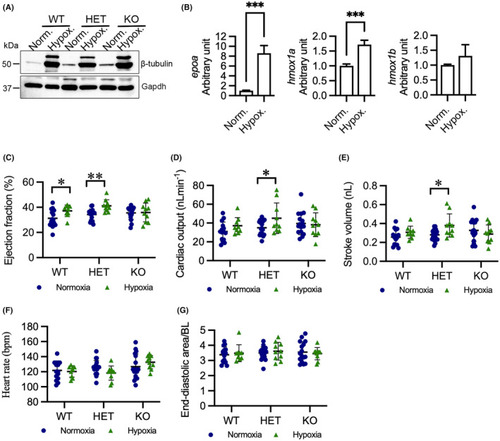
Hypoxia enhances cardiac output in HET(52del) larvae. (A) Representative immunoblots of β‐tubulin and Gapdh in WT, HET(52del), and KO(52del) at normoxic and hypoxic (3% O2 for 24 h) conditions. Gapdh serves as an internal control. Norm. normoxia; Hypox. Hypoxia. (B) RT‐qPCR analyses indicate that hypoxia induces upregulation of |
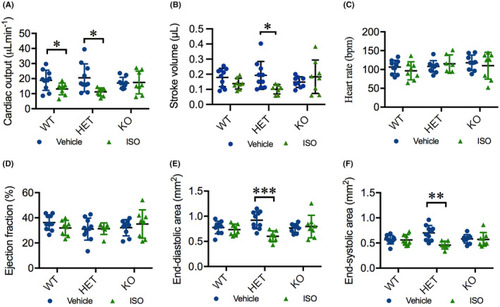
ISO deteriorates cardiac function in HET(52del) adults. (A) ISO treatment results in reduced cardiac output in WT and HET(52del), but not in KO(52del), compared to vehicle‐treated controls. (B) ISO treatment leads to reduced stroke volume in HET(52del), not in WT and KO(52del) compared to vehicle‐treated controls. (C, D) ISO treatment has no significant effect on heart rate (C) or ejection fraction (D) in the three genotypic groups compared to respective vehicle controls. (E, F) ISO treatment leads to reduced end‐diastolic (E) and end‐systolic area (F) in HET(52del), not in WT and KO(52del) compared to vehicle‐treated controls. WT, vehicle |
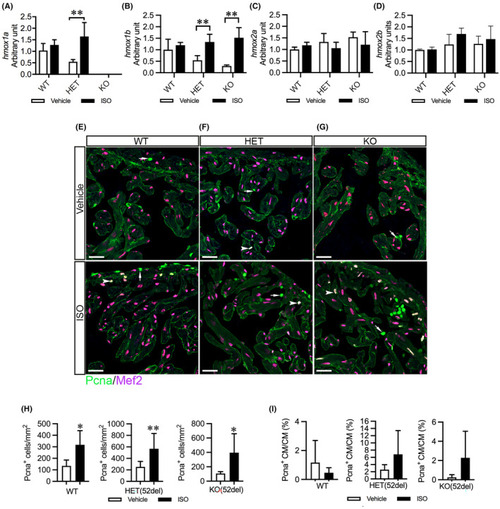
ISO upregulates cardiac |