
- Title
-
Base editing-mediated perturbation of endogenous PKM1/2 splicing facilitates isoform-specific functional analysis in vitro and in vivo
- Authors
- Lin, J., Wu, S., Shen, Q., Liu, J., Huang, S., Peng, G., Qiao, Y.
- Source
- Full text @ Cell Prolif.
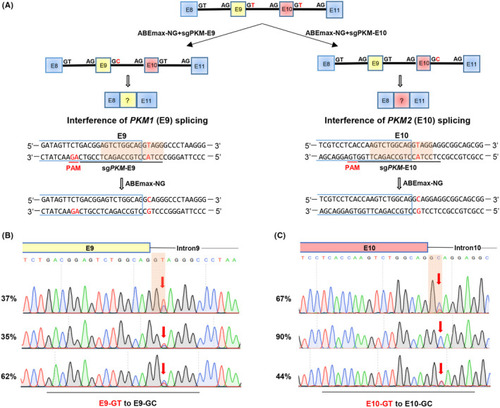
Base editing‐mediated efficient mutation of splicing junction sites of |
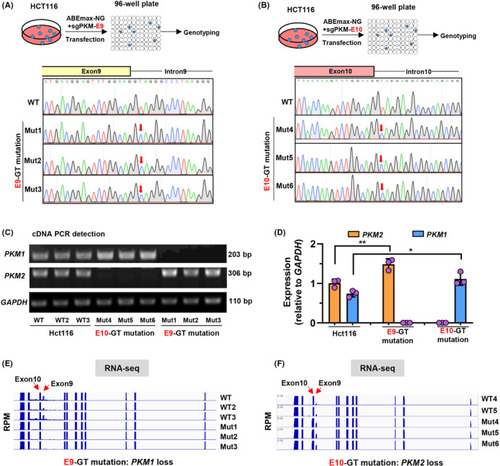
Splicing junction site mutations of |
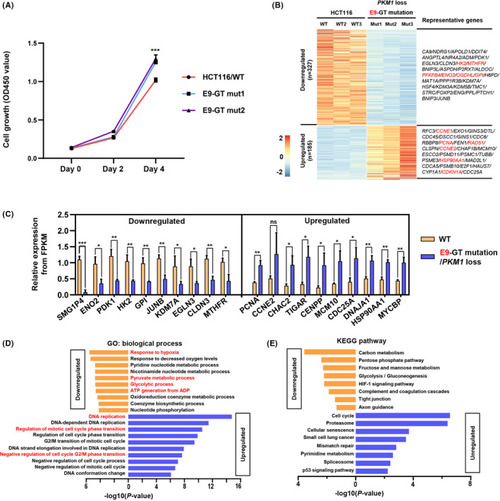
|
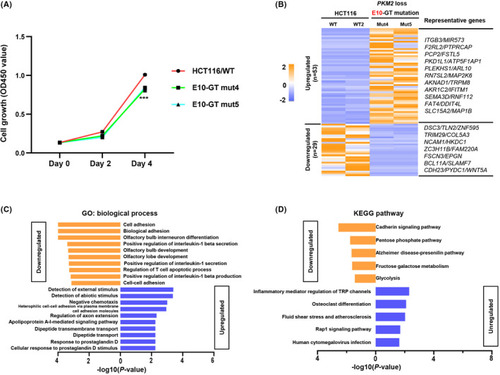
|
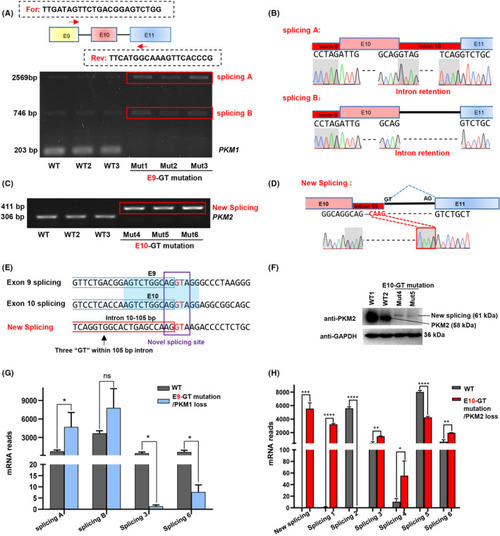
Interference of endogenous |
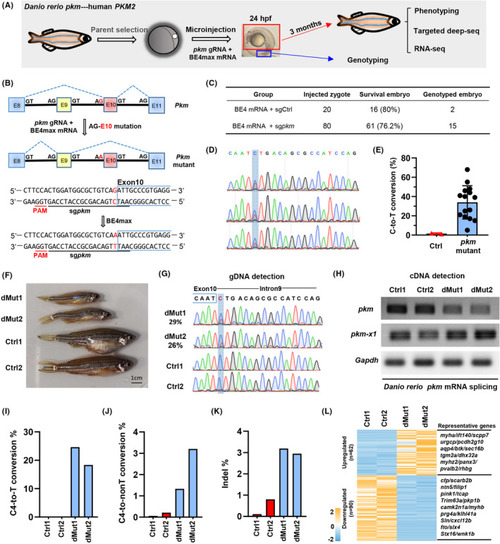
Interference of |

Model for base editing‐mediated splicing perturbation in functional analysis. Because of the lack of appropriate techniques, the specific functions of the 2 |