
- Title
-
Complement System Inhibitory Drugs in a Zebrafish (Danio rerio) Model: Computational Modeling
- Authors
- Fernandes, D.C., Tambourgi, D.V.
- Source
- Full text @ Int. J. Mol. Sci.

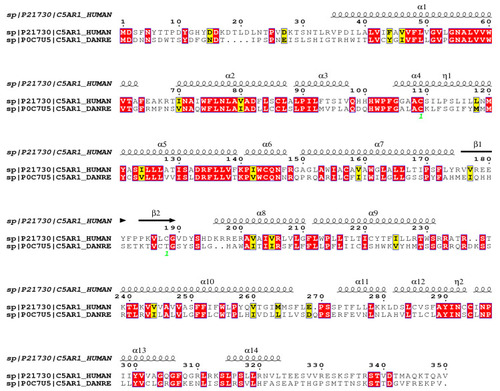
Homology analysis between the β chains of C3 molecules from |
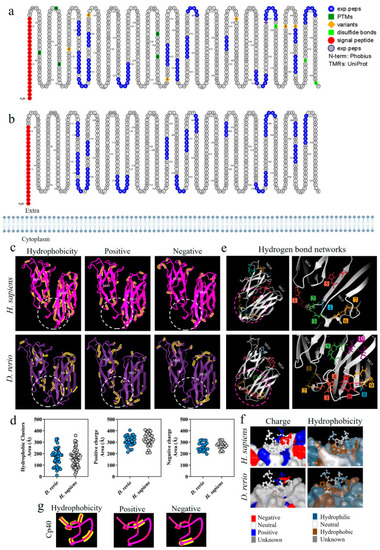
Comparative analysis on the C3-molecule β chains from |
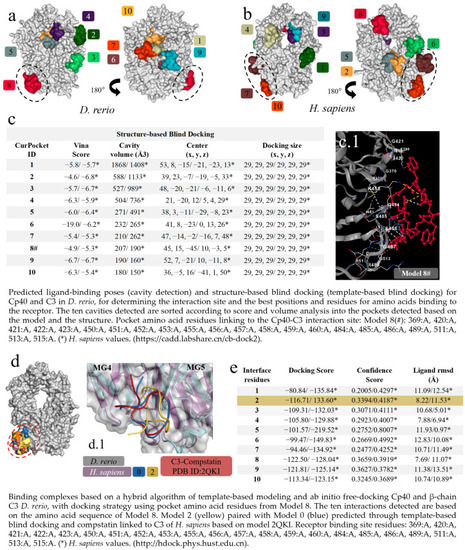
Virtual screening for the detection of the best |
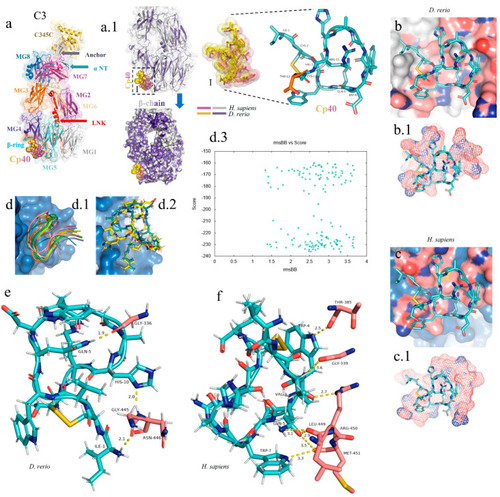
In silico analysis of the peptide–protein interaction between the peptide Cp40 and the C3 molecule of |
Comparative structural alignment between |
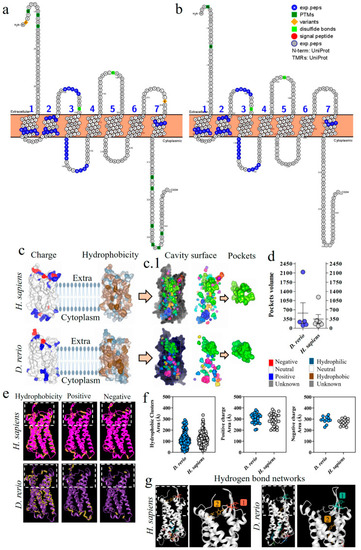
Comparative analysis of C5aR1 from |
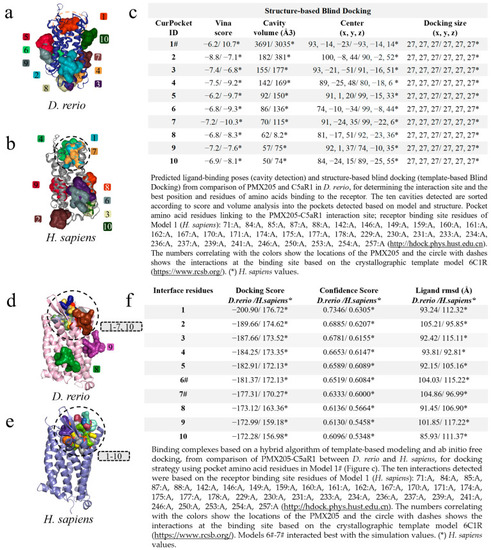
Virtual screening for the detection of the best |
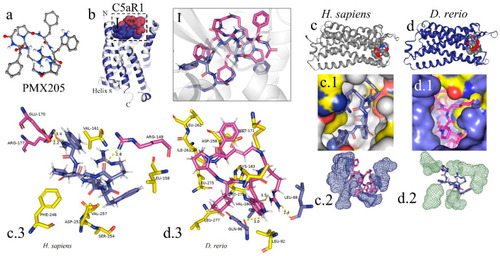
In silico analysis of the interaction of PMX205 with C5aR1 in |