
- Title
-
Comparative Transcriptome Analysis of the Regenerating Zebrafish Telencephalon Unravels a Resource With Key Pathways During Two Early Stages and Activation of Wnt/β-Catenin Signaling at the Early Wound Healing Stage
- Authors
- Demirci, Y., Cucun, G., Poyraz, Y.K., Mohammed, S., Heger, G., Papatheodorou, I., Ozhan, G.
- Source
- Full text @ Front Cell Dev Biol
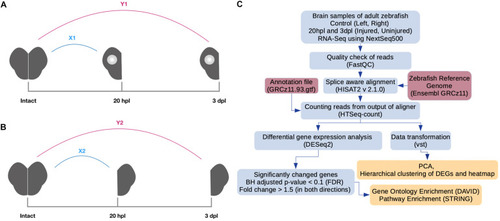
Pipeline for RNA preparation and analysis of RNA sequencing (RNA-seq) data. |
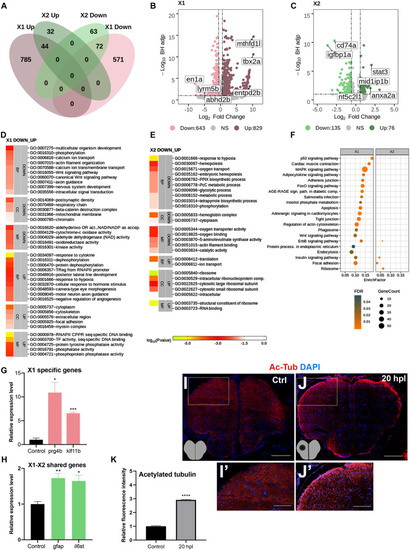
Transcriptome profiling of the telencephalon during the early wound healing stage of regeneration. |
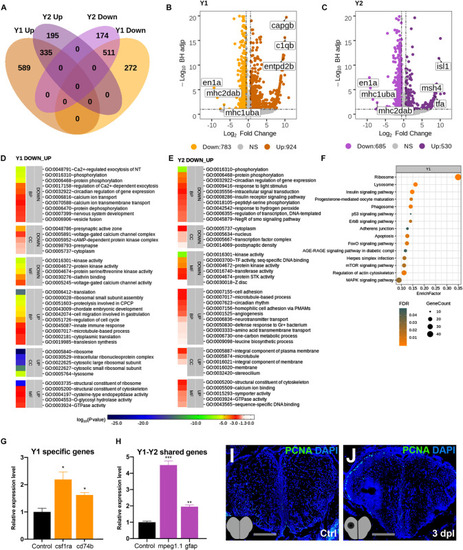
Transcriptome profiling of the telencephalon during the early proliferative stage of regeneration. |
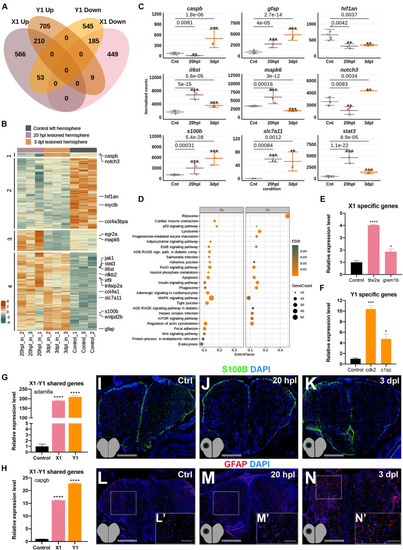
Early wound healing and proliferative stages of brain regeneration are more different than similar. |
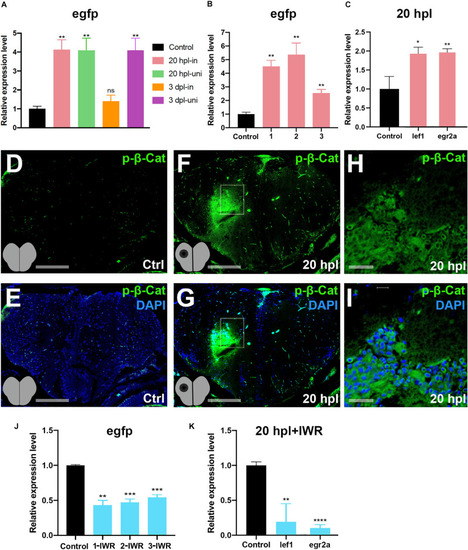
Wnt/β-catenin signaling is activated during the early wound healing stage in the regenerating brain. |
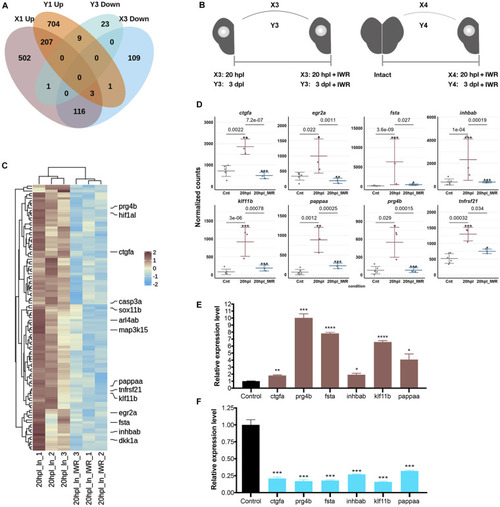
Inhibition of Wnt/β-catenin signaling during the early wound healing stage identifies 119 target genes that are positively regulated by the pathway. |
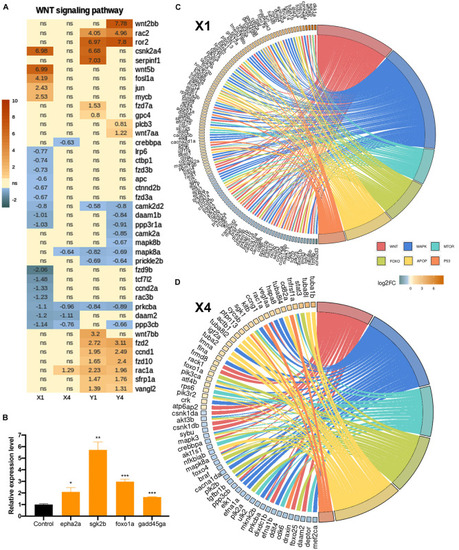
Inhibition of Wnt/β-catenin signaling during the early wound stage results in a marked alteration of the gene expression profiles represented in Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment. Heatmap representation of the uniquely changed genes in |