
- Title
-
DANGER analysis: risk-averse on/off-target assessment for CRISPR editing without a reference genome
- Authors
- Nakamae, K., Bono, H.
- Source
- Full text @ Bioinform Adv
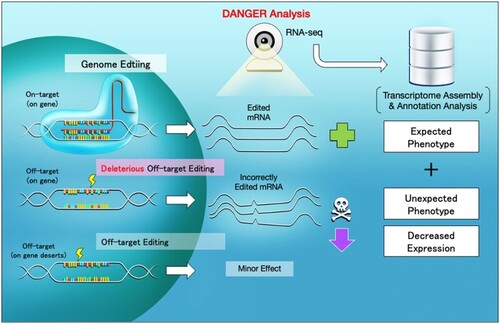
Scheme of CRISPR-Cas9 targeting, deleterious off-target editing, and DANGER analysis. |
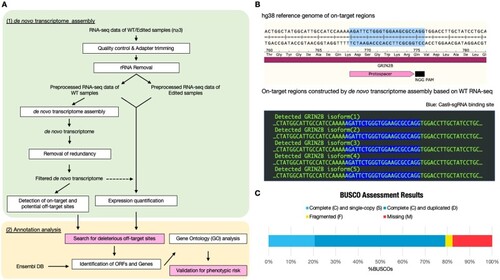
Overview of DANGER analysis and on-target region constructed by |

The benchmark for expression analysis methods compared with reference-based RNA-seq analysis using RNA-seq data derived from WT and GRIN2B edited iPSC-derived cortical neurons. (A) Comparison of different expression analyses. A Venn diagram comparing the |

The result of risk assessment in DANGER analysis using RNA-seq data derived from WT and GRIN2B edited iPSC-derived cortical neurons. (A) An example of the annotation table for DANGER analysis. The table includes GO ID, GO term, number of MMs ( |
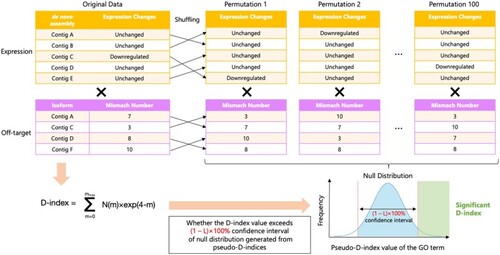
A scheme for permutation testing to evaluate the validity of the D-index. The thin arrow indicates the manipulation of rearranging values from the original expression and off-target profile to the permutation data. The cross represents the computation for applying the D-index formula to the above expression profile and the below off-target profile data. The workflow is shown as the bold arrows. |
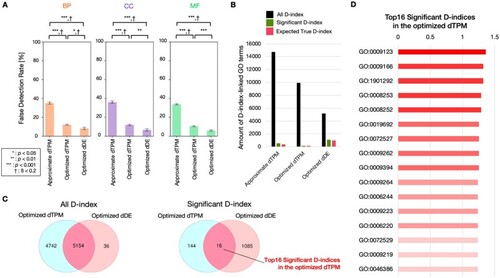
Evaluation of permutation test for DANGER analysis and comparison between dTPM and dDE. (A) Comparison of false detection rates among approximate dTPM (up to 11-MM NRR PAM, |
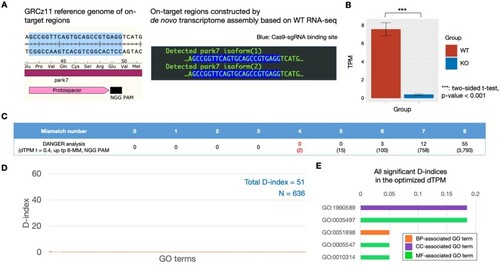
DANGER analysis result using RNA-seq data derived from WT and park7 (dj1) edited brains of |
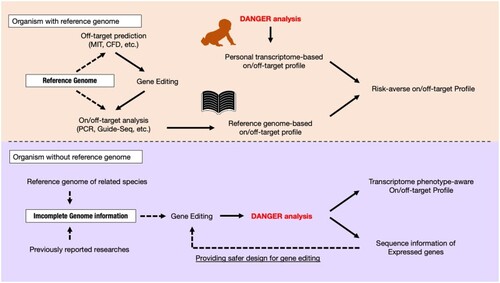
Our proposal for the usage of DANGER analysis in organisms with and without a reference genome. The workflow is shown as black arrows. The dotted black arrows indicate the front of the arrow and refer to the arrow base information. The image of the book is from TogoTV (© 2016 DBCLS TogoTV, CC-BY-4.0, |