
- Title
-
Mutations in Bcl9 and Pygo genes cause congenital heart defects by tissue-specific perturbation of Wnt/β-catenin signaling.
- Authors
- Cantù, C., Felker, A., Zimmerli, D., Prummel, K.D., Cabello, E.M., Chiavacci, E., Méndez-Acevedo, K.M., Kirchgeorg, L., Burger, S., Ripoll, J., Valenta, T., Hausmann, G., Vilain, N., Aguet, M., Burger, A., Panáková, D., Basler, K., Mosimann, C.
- Source
- Full text @ Genes & Dev.
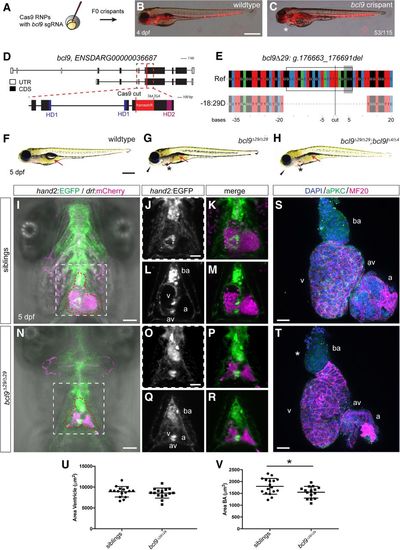
Mutations in bcl9 lead to cardiac defects in zebrafish. (A) A CRISPR–Cas9-mediated mutagenesis identified bcl9 as a potential regulator of heart morphogenesis. (B,C) bcl9 crispants have heart-looping defects, as visible in drl:mCherry transgenics, leading to cardiac edema (asterisk) (lateral view; anterior is to the left). (D) Schematic representation of the bcl9 gene locus and generation of the bcl9Δ29 germline allele. A sgRNA was designed to target the coding exon 6 between HD1 and HD2 of the zebrafish bcl9 gene. The locus is represented as per annotation Zv10, with two isoforms that differ in the first coding exon and the untranslated regions (UTRs). The dotted box represents a zoomed region of the locus, and the red line marks the location of the sgRNA to introduce the Cas9-mediated cut, which resulted in the indicated frameshift (red box) followed by two stop codons in the isolated bcl9Δ29 allele. In the isolated allele, black boxes mark coding exons (CDS), white boxes mark UTRs, blue boxes represent the CDSs that contribute to HD1, and purple boxes represent the CDSs that contribute to HD2. (E) CrispRVariants panel plot depiction of the bcl9Δ29 germline allele with a 29-base-pair (bp) deletion. The top shows genomic reference bcl9Δ29 (bcl9:g.176663_176691), shown below. bcl9Δ29 features an out-of-frame deletion introducing a frameshift followed by 157 novel amino acids terminated by two consecutive stop codons, thus disconnecting HD1 from HD2. The black box indicates the exact position of the sgRNA sequence, the gray-shaded box indicates the 5′-NGG-3′ PAM sequence, and the black line indicates the predicted Cas9-induced double-strand break position. (F–H) Bright-field images of 5-d post-fertilization (dpf) homozygous bcl9Δ29 and bcl9Δ29;bcl9lΔ4 embryos and their wild-type-looking siblings (lateral views; anterior is to the left). Mutant embryos showed heart-looping defects and cardiac edema (asterisks). Moreover, mutant embryos did not inflate their swim bladders (arrows), presumably due to a failure in gasping air because of craniofacial malformations (black arrowheads). (I–R) Single-plane illumination microscopy (SPIM) images of hand2:EGFP;drl:mCherry-expressing wild-type siblings and homozygous bcl9Δ29 embryos (ventral views; anterior is to the top; imaged after viable heart-stopping BDM treatment). I and N depict maximum-intensity projections, and J, K, O, and P show close-ups of the dotted square in I and N, while L, M, Q, and R depict optical sections at the atrio–ventricular canal level. Compared with siblings that form correctly looped hearts with atrio–ventricular canal valves and a bulbus arteriosus (BA; heart outlined with red dotted line; n = 4; J–M), bcl9Δ29 embryos show heart-looping defects (N–R) and craniofacial malformations in both cartilage and head vascularization (n = 8; N). (S–V) Six-micrometer Z-confocal projections of sibling control (S) and homozygous bcl9Δ29 (T) hearts at 5 dpf. Quantification of the ventricular (U) and BA area (V) in sibling hearts (n = 16) compared with homozygous bcl9Δ29 hearts (n = 15) shows that the BA area is significantly smaller in the bcl9Δ29 hearts. (*) P < 0.0221, unpaired t-test with Welch correction. Each data point represents the averaged ventricular or BA area from one heart; quantification was derived from three independent experiments. N = 3. (ba) BA; (a) atrium; (v) ventricle; (av) atrio–ventricular canal. Bars: B,C,F–H, 500 µm; I–R, 100 µm; S,T, 20 µm. |
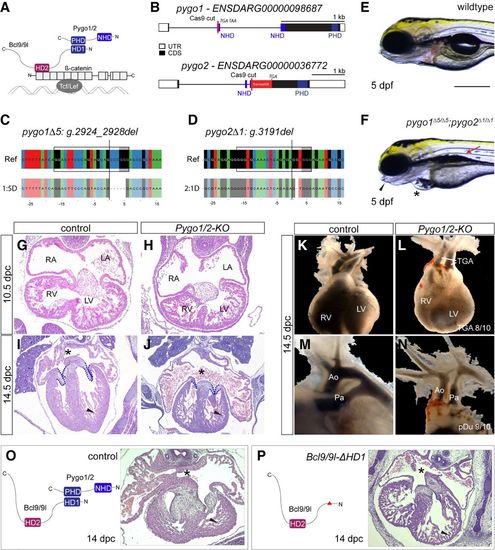
Pygo1/2 mutant zebrafish and mouse embryos develop cardiac malformations reminiscent of CHD. (A) Schematic of the tripartite complex comprised of β-catenin, BCL9, and Pygo with their individual interaction domains together tethered to a WRE by TCF/LEF. (B) Schematic representation of the zebrafish pygo1 and pygo2 genes with annotated NHDs and PHDs and the Cas9 cutting site to generate mutants. The gene locus is represented as per genome annotation Zv10, with the main isoforms of both genes shown. See also the legend for Figure 1B. (C,D) CrispRVariants panel plot depictions of the germline alleles with a 5-bp or 1-bp deletion in pygo1 and pygo2, respectively. The top shows the genomic reference with the pygo1Δ5:g.2924_2928del and pygo2Δ1:g.3191del alleles shown below. Both alleles result in an out-of-frame deletion introducing a frameshift in the CDS. See also the legend for Figure 1C. (E,F) Bright-field images of live pygo1Δ5;pygo2Δ1 double mutants reveal cardiac edema (asterisks), craniofacial defects (arrowheads), and aberrant swim bladder inflation (arrows) as detected in bcl9Δ29 mutants (lateral views; anterior to the left). (G–J) Hematoxylin/eosin-stained transverse sections of a murine heart at 10.5 d post-coitum (dpc) (G,H) and 14.5 dpc (I,J). At 10.5 dpc, the development of the heart and heart cushion was still largely normal in the mutants. At 14.5 dpc, the mutants displayed markedly smaller and thinner valves (dashed reduced compact outline), reduced compact ventricular myocardium (arrowhead), highly dilated atria, and defective or absent atrial septum (asterisks). (RA) Right atrium; (LA) left atrium; (RV) right ventricle; (LV) left ventricle. (K–N) Gross anatomical view of the heart and great vessels at 13.5/14.5 dpc as revealed by India ink injection. While normally the aorta (Ao) arises from the left ventricle (LV), and the pulmonary artery (Pa) arises from the right ventricle (RV), mutants showed a classic transposition of the great arteries (TGA; arrows) and hypoplastic aorta and pulmonary artery. (O,P) Schematic representation of the Bcl9/9l–Pygo1/2 interaction (O), the molecular configuration of this interaction when the HD1 domain in Bcl9/9l is deleted in Bcl9/9l-ΔHD1 mice (P), and heart sections at 14 dpc, stained with hematoxylin/eosin. The abrogation of this interaction leads to a delayed chamber septation (asterisks), hypoplastic myocardium (arrowhead), and valve deficiency. Bars: E,F, 250 µm. PHENOTYPE:
|
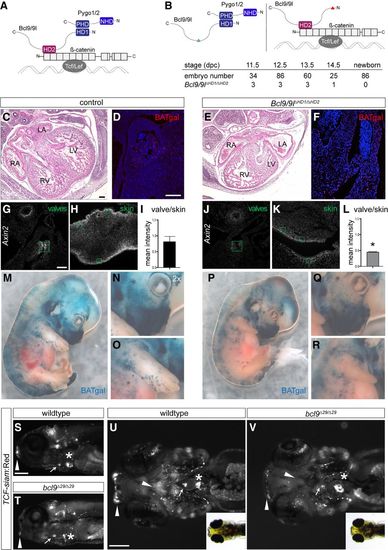
The BCL9–Pygo complex drives Wnt/β-catenin transcription during heart development. (A) Schematic representation of the β-catenin transcriptional complex to outline the experimental strategy. BCL9/9l simultaneously interacts with Pygo1/2 and β-catenin via the HD1 and HD2, respectively. (B) In Bcl9/9l-Δ1/Δ2 mice, no Bcl9/9l molecule can simultaneously bind Pygo1/2 and β-catenin; the deletion of the HD2 or HD1 abrogates Bcl9/9l binding to β-catenin (left panel) or Pygo1/2 (right panel), respectively. Bcl9/9l-Δ1/Δ2 embryos are never found after 14.5 dpc (table). (C–F) Hematoxylin/eosin staining on heart sections of control (C) and Bcl9/9l-Δ1/Δ2 (E) embryos at 13.5 dpc. Bcl9/9l-Δ1/Δ2 embryos have pronounced heart defects, with thinned myocardium of the ventricular walls and malformations of the forming septum, the atrio–ventricular valves, and OFT. The expression of in vivo BATgal Wnt reporter in the OFT region is markedly reduced in Bcl9/9l-Δ1/Δ2 mutant (F) compared with control (D) hearts. (RA) Right atrium; (LA) left atrium; (RV) right ventricle; (LV) left ventricle. (G–L) Axin2 expression (detected by single-molecule mRNA ISH) in wild-type and Bcl9/9l-Δ1/Δ2 OFT valves (G,J) and skin (H,K) at 13.5 dpc. Green boxes are drawn around transversal sections of individual valves of the OFT (G,J) and in skin sections from the limbs (H,K) for quantification of fluorescence intensity levels (I,L). Axin2 expression is strongly reduced in the valves of Bcl9/9l-Δ1/Δ2 embryos (J), while reduction in the skin is mild (K). (I,L) The quantification of the ratio between Axin2 expression in the valves and the skin in the different genotypes reveals a significant reduction of Axin2 expression in Bcl9/9l-Δ1/Δ2 valves. (*) P-value < 0.05. (M–R) In vivo BATgal reporter expression in 13.5-dpc wild-type (M–O) and Bcl9/9l-Δ1/Δ2 mutant (P–R) embryos. Mutant embryos have a slightly decreased reporter expression, but BATgal reporter expression is generally retained in most tissues. Reduced BATgal activity is observed in the craniofacial region (N,Q) and the forelimbs (O,R). Loss of canonical Wnt signaling transcription in the forelimbs is accompanied by severe developmental limb defects in Bcl9/9l-Δ1/Δ2 embryos (cf. O and R). (S–V) Fluorescent and bright-field images of TCF-siam:Red bcl9Δ29 and wild-type zebrafish embryo lateral (S,T) and ventral (U,V) views; anterior is to the left. TCF reporter activity is specifically reduced in the cardiac OFT (arrow) and craniofacial apparatus (arrowheads) and altered in the atrio–ventricular valve (asterisks). Bars: C–F,G,H,J,K, 100 µm; S–V, 200 µm. |
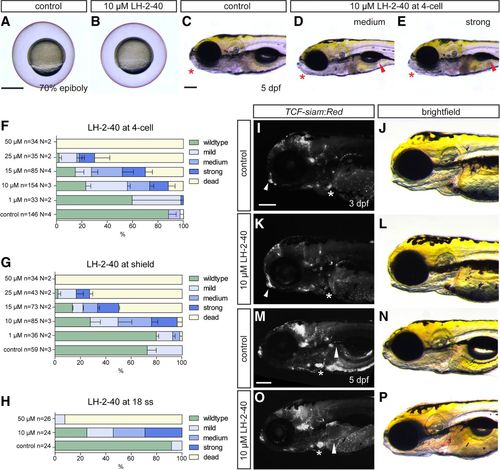
Chemical manipulation of zebrafish heart development by Bcl9–β-catenin inhibitor LH-2-40. (A,B) Treatment with 10 µM LH-2-40 from the two- to four-cell stages does not result in gastrulation defects (lateral views). (C–E) Bright-field images of 5-dpf DMSO controls (C) and representative embryos treated with 10 µM LH-2-40 from the four-cell stage on (D,E), as assessed for phenotype classes (quantified in F–H) (lateral views; anterior is to the left). Phenotypes in LH-2-40-treated embryos become visible at 5 dpf, comparable with phenotypes observed in bcl9Δ29 mutants. LH-2-40-treated embryos show a variable phenotype expressivity, with mild to strong swim bladder inflation defects (arrowheads; D,E) linked with variable craniofacial defects (asterisks; D,E, cf. control in C). (F–J) Similar dose-dependent phenotype penetrance and expressivity are observed after Bcl9 inhibition at the four-cell, shield, and 18-somite (18 ss) stages, suggesting that the observed phenotypes result from perturbed Bcl9 function in craniofacial and heart development after somitogenesis. Mild phenotypes are characterized by mild craniofacial and swim bladder inflation defects. (D) Both phenotypes are more pronounced in the medium phenotype class. (E) Strong phenotypes are characterized by strong craniofacial defects and a complete failure to inflate the swim bladder. (I–P) Fluorescent and bright-field images of TCF:siamRed Bcl9-inhibited (K,L,O,P) and DMSO-treated (I,J,M,N) wild-type siblings at 3 and 5 dpf (lateral views; anterior is to the left). TCF reporter activity is severely reduced in the atrio–ventricular valve at 3 dpf (asterisks; I,K) and altered in the craniofacial cartilage (arrowheads; I,K). At 5 dpf, the TCF reporter is aberrantly patterned in the atrio–ventricular valve (asterisks; M,O) and fins (arrowheads; M,O). Bars: C–E,I–P, 200 µm; A,B, 500 µm. EXPRESSION / LABELING:
PHENOTYPE:
|
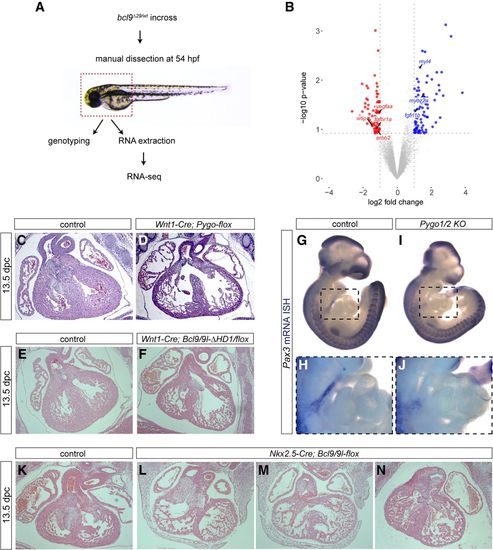
The Bcl9–Pygo complex acts in neural crest and cardiac heart progenitor cells. (A) Schematic representation of the RNA sequencing (RNA-seq) approach used for bcl9Δ29 mutant zebrafish embryos. RNA extraction was performed on manually dissected anterior structures of 54-hpf embryos as indicated by the dashed box. (B) Comparing three independent biological replicates revealed 157 differentially expressed (74 up-regulated and 83 down-regulated) genes between wild-type and bcl9Δ29 mutants as represented in the volcano plot. Genes associated with cardiac development are highlighted. (C–F) Hematoxylin/eosin staining of heart sections of control (C,E) compared with Wnt1-Cre; Pygo1/2-flox (D) and Bcl9/9l-ΔHD1/flox (F) 13.5-dpc siblings. (G–J) Pax3 expression in 10.5-dpc embryos in the region of branchial arches: view of whole-mount mRNA ISH of control (G) compared with Pygo1/2 knockout (KO; I) siblings. H and J are magnified insets of the regions marked with dashed squares in G and I, respectively. (K–N) Hematoxylin/eosin staining of heart sections of control (K) compared with Nkx2.5-Cre; Bcl9/9l-flox (L–N) 13.5-dpc siblings. |
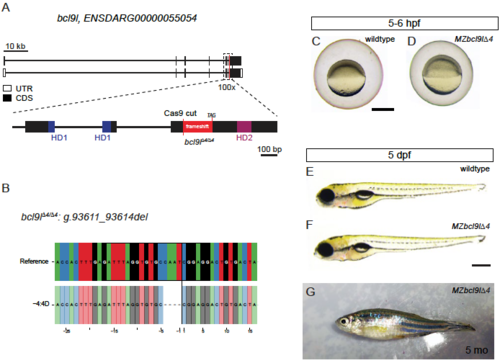
MZbcl9l∆4 mutants are viable and fertile. (A,B) Schematics showing the mutation induced in bcl9l∆4 mutants. Analogous to bcl9 mutants (see Figure 1B,C), we designed a sgRNA that induces mutagenesis between the HD1 and HD2 domain. Gene locus represented as per genome annotation Zv10 (A) with two isoforms that differ in the first coding exon and the untranslated regions (UTRs). The green dotted box represents a zoomed region of the gene locus, with the red line representing the location of the sgRNA used for mutagenesis; black boxes mark coding exons (CDS), white boxes mark UTRs, the blue boxes represent the part of the CDS that will contribute to HD1 and purple boxes to HD2. A 4 bp deletion at the Cas9 cutting side (B) leads to a frame-shift allele with a premature STOP codon before HD2. (C-G) Brightfield images of homozygous bcl9l∆4 mutants at different stages from gastrulation to adulthood, lateral views, anterior to the left. Maternal zygotic bcl9l mutants (MZbcl9l∆4) mutants did not show any gastrulation defects (C,D). At 5 dpf, we could not detect any cardiac and craniofacial phenotypes (E,F) as observed in zygotic bcl9∆29 mutants (see Figure 1). (G) Representative image of a five-month old (5 mo) MZbcl9l∆4 F4 mutant obtained from a cross of two adult homozygous bcl9l∆4 mutants. Scale bars, 500 µm. |
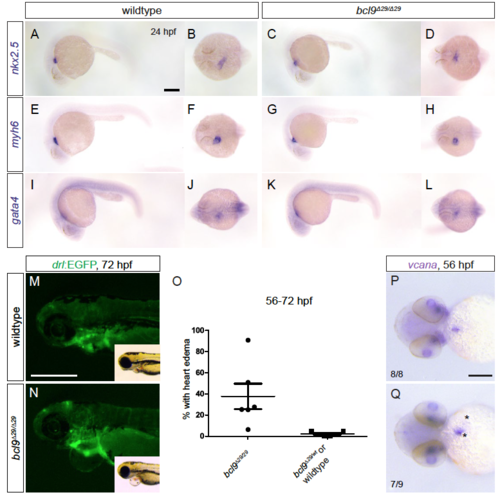
bcl9 mutants feature normal early cardiac patterning, but variable cardiac phenotypes in bcl9Δ29 mutants between 2-3 dpf. (A-L) bcl9 mutants displayed unchanged nkx2.5 (A-D), myh6/amhc (E-H), and gata4 (I-L) expression at 24 hpf compared to wildtype siblings: lateral and dorsal views, anterior to the left. Scale bar, 250 µm. (M-O) Fluorescent and brightfield (inlets) images of drl:EGFP transgenic wildtype and bcl9Δ29 mutant embryos at 72 hpf (M,N), lateral views, anterior to the left. The hearts of bcl9Δ29 are patterned into atria and ventricle. The two chambers are slightly misaligned and the embryos develop cardiac edema (O). (O,P) Whole-mount ISH for vcana reveals expanded expression around the atrio-ventricular canal and in the atrium (asterisks) suggesting a defect in valve formation, ventral views, anterior to the left. Scale bars, 200 µm (M,N), 500 µm (P,Q). Diagram in O depicts mean with s.d. from representative clutch of embryos, genotype confirmed by PCR. |
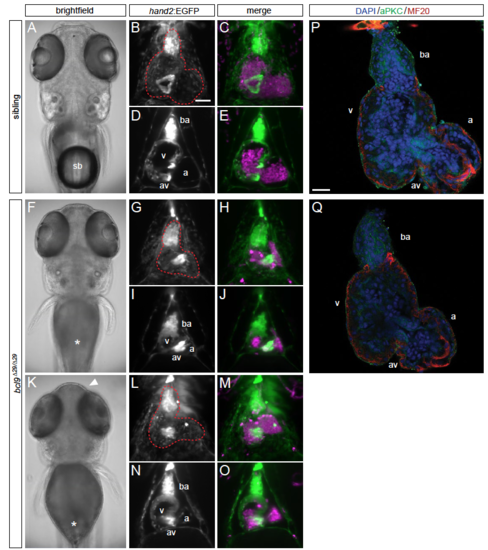
Cardiac phenotypes of zebrafish bcl9Δ29 mutants. (A-O) SPIM images of hand2:EGFP;drl:mCherry-expressing wildtype siblings and homozygous bcl9Δ29 embryos at 5 dpf; ventral views, anterior to the top, imaged after viable heart-stopping BDM treatment, two individual bcl9Δ29-mutant embryos are shown. A,F,K, bright field views to illustrate the absent swimbladder in mutants (sb, asterisks in F,K) and different expressivity of craniofacial defects (arrowhead, K); B-O maximum-intensity projection fluorescence close-ups of the heart (red dotted outlines in B,G,L); D,E,I,J,N,O depict optical sections at AV canal level. (P,Q) Confocal sections of sibling control (P) and homozygous bcl9Δ29 (Q) hearts, dissected and stained at 5 dpf to reveal the details of cardiac architecture. Also compare to Figure 2. ba, bulbus arteriosus; a, atrium; v ventricle; av, atrioventricular canal. Scale bars B-E,G-J,L-O 100 m, S,T 20 m. |

Craniofacial defects in bcl9 and pygo1/2 mutants. (A-F) Alcian blue staining of the pharyngeal cartilage of 5 dpf wildtype, bcl9Δ29, and double homozygous pygo1Δ5;pygo2Δ1 embryos shown in ventral (A,C,E) and lateral (B,D,F) views, anterior to the left. bcl9 and pygo1/2 mutants have severe malformations of the pharyngeal apparatus with fusions defect of the ceratohyal (ch) and ceratobranchial 1 (cb1) arches and miss-shaped Meckel's (m) and palatoquadrate (pq) cartilage. Scale bars, 100 µm. (G-L) SPIM-based brightfield imaging of wildtype-appearing siblings and homozygous pygo1Δ5;pygo2Δ1 embryos. Note absence of swim bladder (sb) and craniofacial defects in double-mutants (G,J). Ventral close-up of cardiac region (square in H,K, enlarged in I,L with red outline depicting the heart), showing abnormal heart looping and smaller bulbus arteriosus region in homozygous mutants. (M) Quantification of phenotypes in four individual pygo1Δ5/wt x pygo2Δ1/wt crosses reveal defects in 7-18% of all embryos. Genotyping of phenotypic embryos revealed phenotype occurrence in homozygous pygo2Δ1 mutants in combination with homozygous or heterozygous pygo1Δ5 mutation. (N-P) MZpygo1Δ5 (N) and homozygous pygo2Δ1 (O) mutants, as well as mutants carrying a homozygous pygo1Δ5 combined with heterozygous pygo2Δ1 alleles (P) are viable and fertile; lateral views, anterior to the left. |
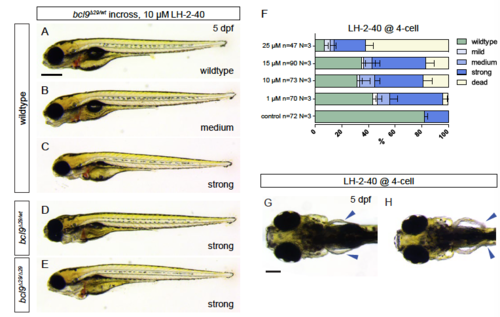
Functional inhibition of Bcl9- -catenin-interaction in bcl9∆29 mutants and wildtype leads to craniofacial, cardiac, and fin defects. (A-E) Embryos obtained from a heterozygous bcl9∆29 (bcl9∆29/wt) incross were treated with 10 µM LH-2-40 from 4-cell stage on, lateral views, anterior to the left. Phenotype classes as described in Figure 4 were observed independent of the genotype of the embryo. (F) General phenotype penetrance is comparable to wildtype embryos treated with 10 µM LH-2-40 from 4-cell stage on (see Figure 4F). Nevertheless, the penetrance of strong phenotypes is increased in treated bcl9∆29 crosses. (G,H) In addition to cardiac and craniofacial defects, Bcl9-inhibited embryos (wildtype or bcl9∆29 incrosses) are characterized by readily observable fin defects not observed in untreated bcl9∆29 mutants (arrow heads H, compare to control in G). |