
- Title
-
Phenotype to genotype: A new and rapid approach using whole-genome sequencing
- Authors
- Feltes, M., Zimin, A.V., Angel, S., Pansari, N., Hensley, M.R., Anderson, J.L., Shen, M.C., Klemek, M., Shen, Y., Ginde, V.S., Kozan, H., Le, N.V., Truong, V.P., Wilson, M.H., Salzberg, S.L., Farber, S.A.
- Source
- Full text @ PLoS Genet.
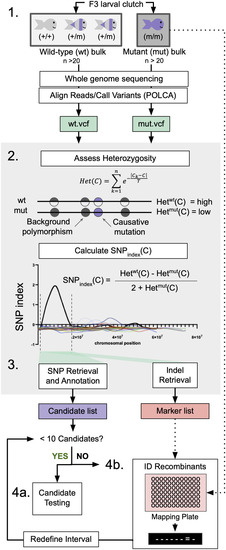
WheresWalker pipeline utilizes WGS data to identify segregating SNPs and indels. 1. Bulk Segregant Analysis: animals are sorted by phenotype and pooled to generate wild-type (wt) and mutant (mut) genomic DNA for whole-genome sequencing. Sequencing data is aligned and evaluated for variance using POLCA which outputs VCF files for wt and mut samples. gDNA for additional mutant animals can be saved for downstream analyses. 2. Heterozygosity is calculated in a sliding window across wt and mut genomes, where C is the coordinate at the center of each 10,000 bp window. These values are used to calculate the SNP index in order to define a homozygous interval; dashed lines indicate interval bounds. 3. Whereswalker extracts SNPs and indels that segregate appropriately with the mutant phenotype to generate a list of candidate SNPs and a list of indel markers. Steps 2 and 3 are executed in a single command by the WheresWalker script. 4a. If sufficiently few candidates have been identified, the genes can be targeted with CRISPR/Cas9. 4b. If the number of targets is intractable, the interval can be refined by identifying recombinants. This can be repeated until a sufficiently short candidate list has been generated. |
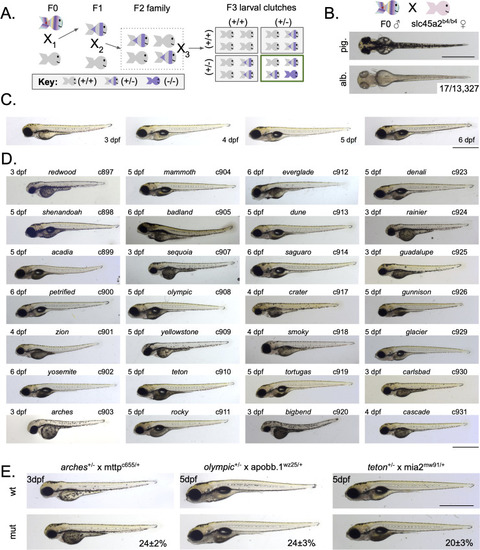
Forward genetic screen identifies 28 dark yolk mutants. A) Generation of mutant families using a standard forward genetic F3 screening scheme. B) Single locus hit rate for the |
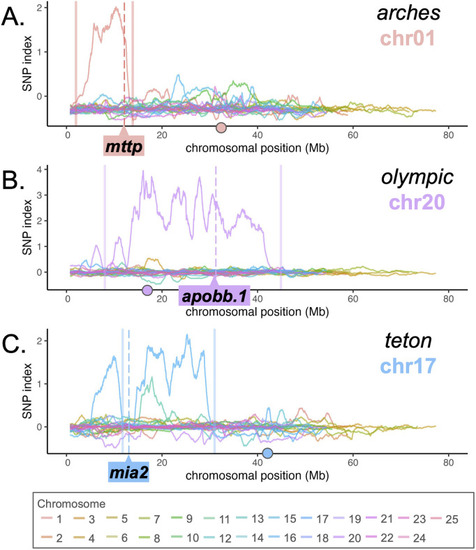
WheresWalker identifies the correct chromosomal region for three dark yolk loci. Profile of SNP index across all chromosomes for |
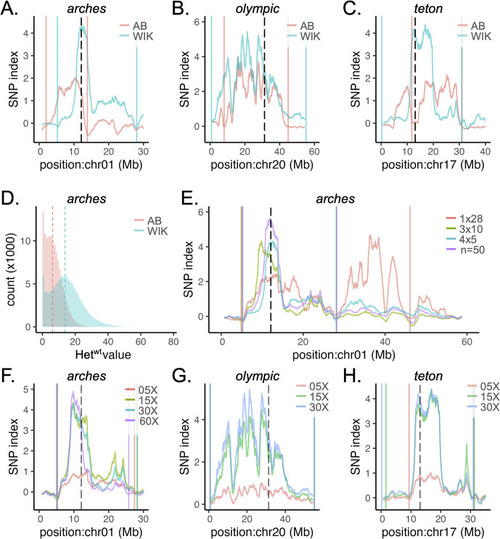
Background heterogeneity and sequencing depth improve WheresWalker SNPindex. A-C) regional SNP index profile for datasets generated in an AB or WIK background for |
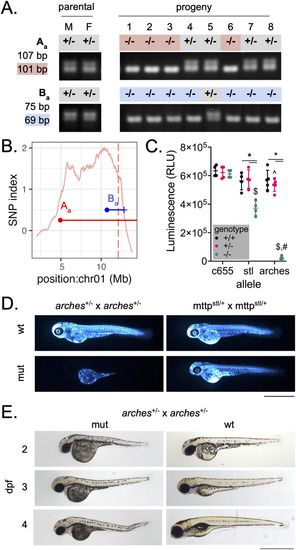
Recombinant mapping narrows region of interest to identify a loss-of-function allele of A) markers Aa and Ba were outputted by WheresWalker and used to genotype |
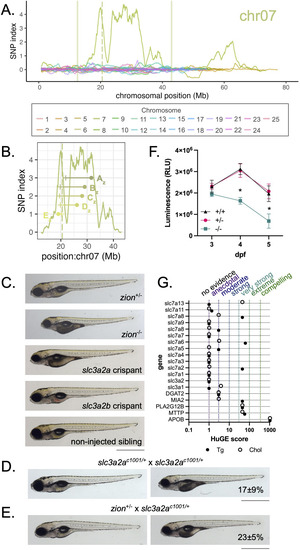
A) Elevated SNP index is observed on chromosome 7 in |