
- Title
-
PKHD1L1, a gene involved in the stereocilia coat, causes autosomal recessive nonsyndromic hearing loss
- Authors
- Redfield, S.E., De-la-Torre, P., Zamani, M., Wang, H., Khan, H., Morris, T., Shariati, G., Karimi, M., Kenna, M.A., Seo, G.H., Xu, H., Lu, W., Naz, S., Galehdari, H., Indzhykulian, A.A., Shearer, A.E., Vona, B.
- Source
- Full text @ Hum. Genet.
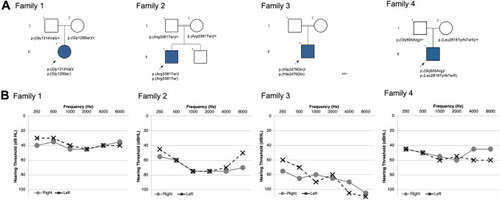
Pedigrees and audiograms. Pedigree and audiometric information for four families with biallelic |
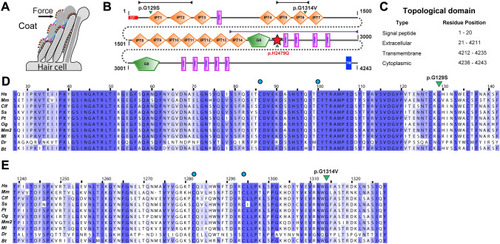
PKHD1L1 protein domain prediction and evolutionary analysis for missense variants (Family 1 and Family 3). |
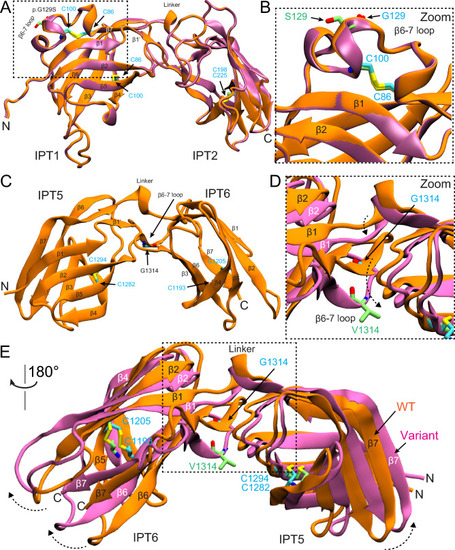
AlphaFold2 modeling of PKHD1L1 protein fragments carrying p.(Gly120Ser) and p.(Gly1314Val) mutations. |
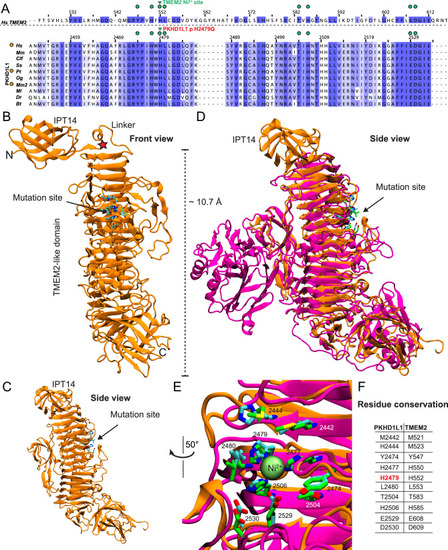
PKHD1L1 structural modeling of the protein fragment containing the p.(His2479Gln) variant. Based on AlphaFold2 predictions, this fragment of PKHD1L1 shares a common fold with the TMEM2 protein within the region carrying the p.(His2479Gln) variant. |
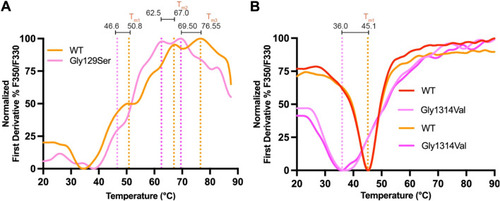
Thermodynamic and folding stability evaluation of two missense variants using NanoDSF. |
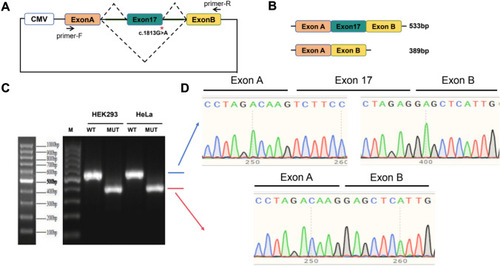
Minigene splicing assay for evaluation of the functional effect of p.(Gly605Arg) on splicing. |