|
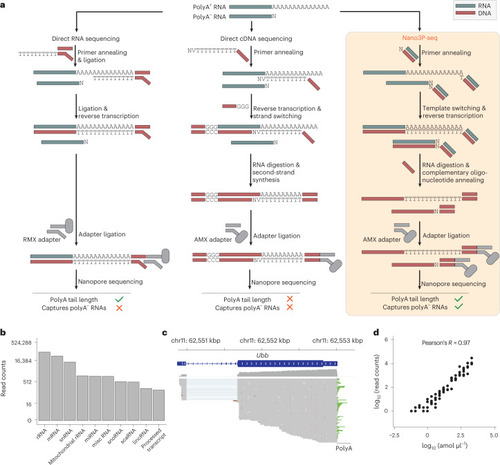
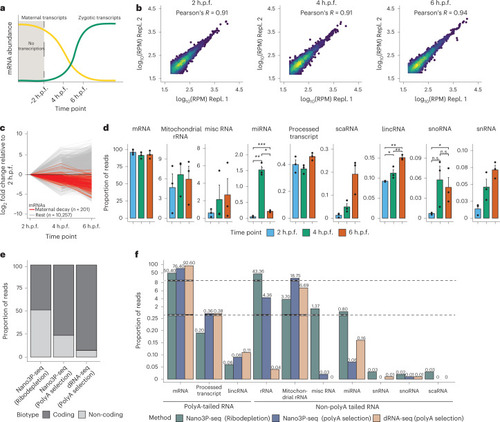
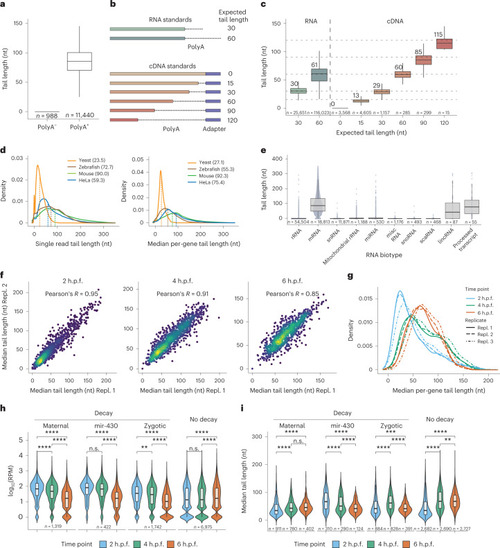
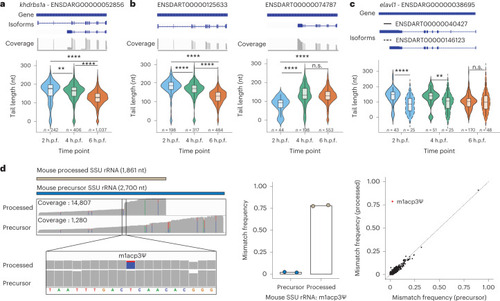
Nano3P-seq can be used to accurately estimate polyA tail lengths in individual molecules.a, PolyA tail length estimates of non-polyadenylated (curlcake 1) and polyadenylated (curlcake 2) synthetic RNAs sequenced with Nano3P-seq. See also Extended Data Fig. 1a–c. nt, nucleotides. b, Schematic overview of the standards used to assess the tail length estimation accuracy of Nano3P-seq. c, Box plots depicting tail length estimations of RNA and cDNA standards sequenced with Nano3P-seq. Values on box plots indicate the median polyA tail length estimation for each standard. d, PolyA tail length distribution of yeast, zebrafish, and mouse mRNAs represented as single-transcript values (left) and per-gene medians (right). e, PolyA tail length estimates across different RNA biotypes from mouse brain total RNA enriched in nuclear/mitochondrial RNA. Each dot represents a read. f, Replicability of median per-gene polyA tail length estimations of zebrafish embryonic mRNAs between two biological replicates for three different time points (2, 4, and 6 h.p.f.). g, Median per-gene polyA tail length distribution of zebrafish embryonic mRNAs across zebrafish developmental stages (2, 4, and 6 hpf, shown in blue, green, and red, respectively) in three biological replicates (shown as full lines, dashed lines, and dotted/dashed lines, respectively). h, Comparative analysis of mRNA abundances (shown as log10(RPM) counts) of zebrafish mRNAs binned according to their annotated decay mode (maternal decay, zygotic activation-dependent decay, miR-430-dependent decay, and no decay) during early embryogenesis (t = 2, 4, and 6 h.p.f.). i, Median per-gene polyA tail length estimations of zebrafish mRNAs binned according to their decay mode (maternal, miR-430, zygotic, and no decay) at 2, 4, and 6 h.p.f. For Fig. 3h,i; statistical analyses were performed using the Kruskal–Wallis test. c,e,h,i, The number of observations included in the analysis is shown below each box and violin plot. Box plot limits are defined by lower (bottom) and upper (top) quartiles. The bar indicates the median, and whiskers indicate ±1.5× interquartile range. Source data
|