
- Title
-
Deploying MMEJ using MENdel in precision gene editing applications for gene therapy and functional genomics
- Authors
- Martínez-Gálvez, G., Joshi, P., Friedberg, I., Manduca, A., Ekker, S.C.
- Source
- Full text @ Nucleic Acids Res.

MENTHU predicts which DNA double-strand breaks likely result in single majority deletions. ( |
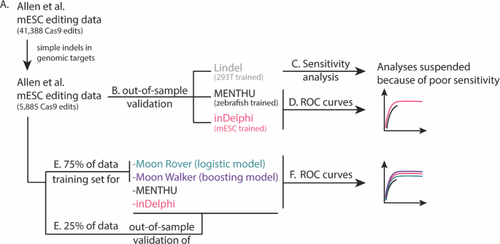
Workflow of the independent assessment of the ability of MENTHU to predict PreMAs. ( |
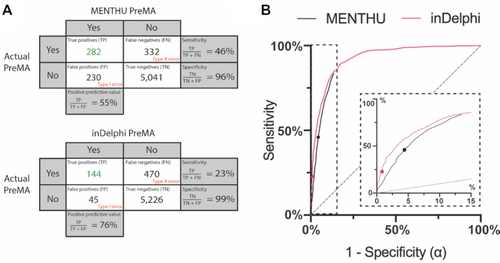
Comparison of the performance of the published versions of MENTHU and inDelphi in predicting PreMAs in a large, out-of-sample dataset. ( |
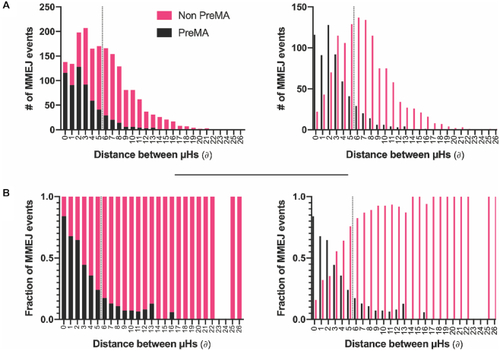
PreMA distribution of MMEJ events as a function of the distance between the microhomologies employed for repair. ( |

Receiver Operating Characteristic (ROC) curves comparing the prediction performance of MENTHU and inDelphi to that of the novel MENTHU-based tools Moon Rover and Moon Walker. Moon Walker and Moon Rover are two machine-learning-based tools that utilize the same two features for PreMA predictions that MENTHU uses: the MENTHU Score and the distance between the microhomologies used for most expected MMEJ repair outcome. The ROC curves displayed represent the PreMA prediction performance of MENTHU, inDelphi, Moon Rover, and Moon Walker on the out-of-sample validation set described on Figure |
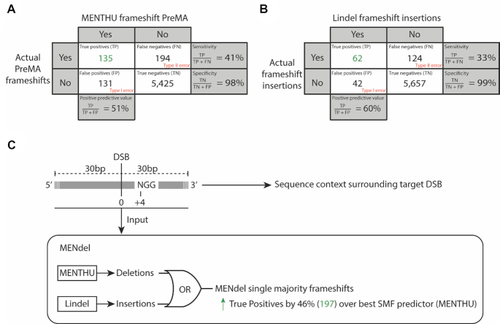
MENdel predicts which DNA double-strand breaks likely result in single majority deletions and insertions for likely frameshift loss of function alleles. The confusion matrices display the performance of the prediction of ( |
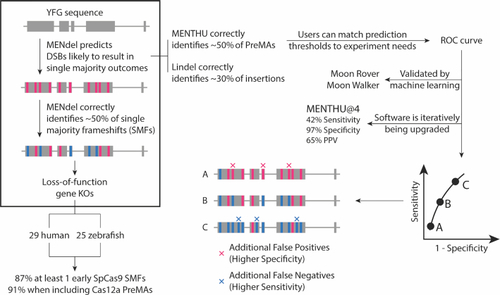
MMEJ-targeting of double-strand break sites for functional genomics and gene therapies. MENdel provides genome engineers with the largest prediction coverage of single majority frameshifts for loss-of-function experiment design (boxed). We sampled 54 vertebrate genes for knockout-generating PreMAs using MENTHU and MENdel, and estimated that the majority (∼90%) of vertebrate genes should possess at least one early out-of-frame single majority outcome. MENTHU (right) is the only double-strand break repair prediction algorithm that allows DNA targeting with nucleases different to SpCas9 and offers scientists with customizable prediction thresholds to best accommodate user needs. |