
- Title
-
The lasting after-effects of an ancient polyploidy on the genomes of teleosts
- Authors
- Conant, G.C.
- Source
- Full text @ PLoS One
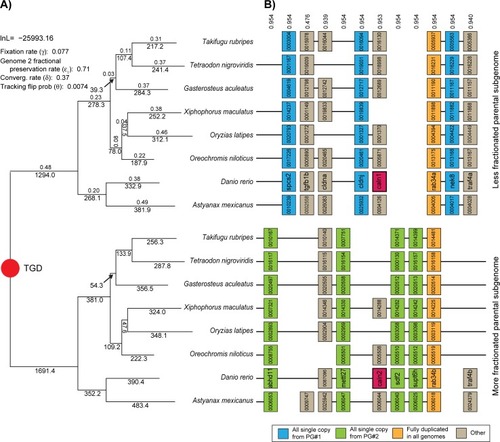
|
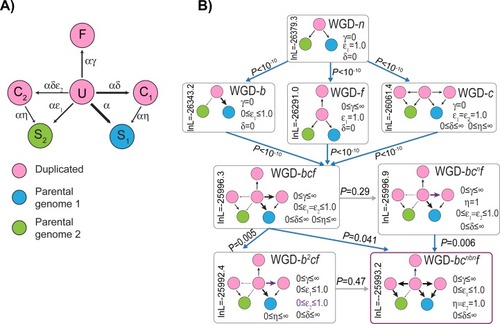
|
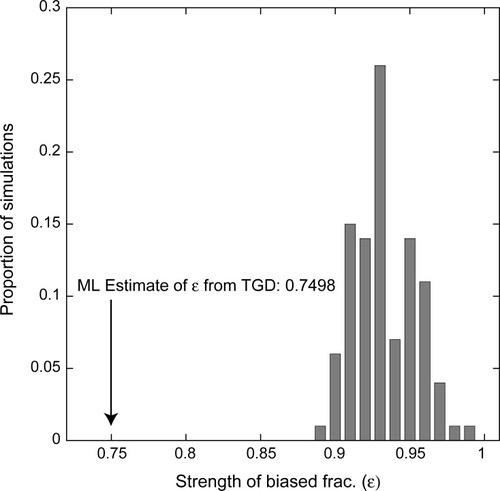
Estimates of ε from these 100 simulations are always less than 1.0 because the model fits stochastic variations in the preservation patterns as potential biased fractionation. However, this stochastic variation never yields estimates of ε as small as seen in the real dataset ( |
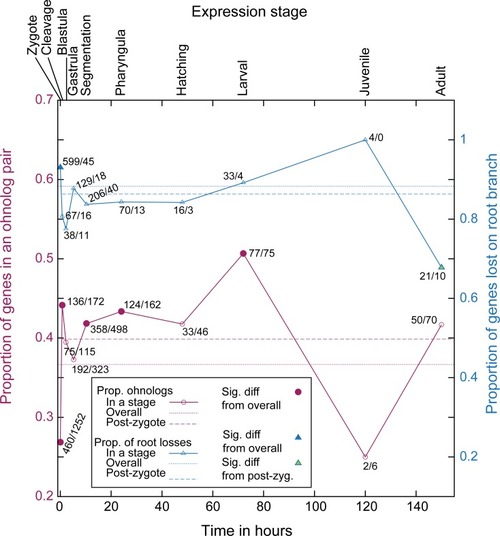
On the |