|
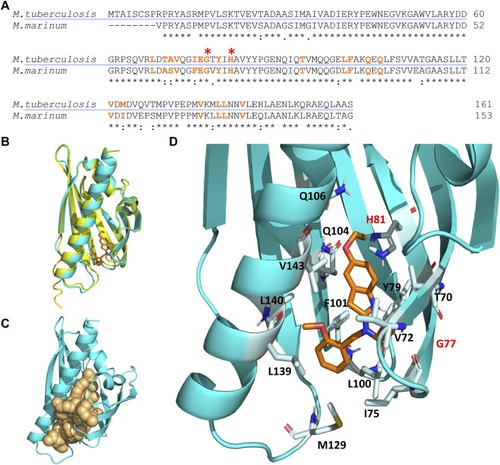
Molecular docking simulation of the structure of the Rv0164 complex with BT-37. (A) Protein sequence alignment of MMAR_0407 in M. marinum and Rv0164 in M. tuberculosis using Clustal Omega. An asterisk (*) indicates positions that have identical residues, a colon (:) indicates that the residues have conserved properties, and a dot (.) indicates residues that are semi-conserved. Amino acids highlighted in orange are in 2 Å vicinity of the compound BT-37 and contribute to inhibitor binding. Red asterisks indicate residues contributing to experimentally confirmed resistance in M. marinum (Mmar G69/Mtb G77 and Mmar H73/Mtb H81). (B) Structure alignment of MMAR_0407 in M. marinum (yellow) and Rv0164 in M. tuberculosis (cyan). The RMSD score between these structures is 0.158 Å. In orange is depicted the compound BT-37 as the top model of docking simulation, according to the HADDOCK score. (C) Definition of the top-scored binding pocket in Rv0164 that was used to define the binding information of the docking simulation. (D) Top model of the docking simulation, according to the HADDOCK score, is shown in more detail. The corresponding residues in M. tuberculosis (G77, H81) contributing to experimentally confirmed resistance in M. marinum are labeled in red.
|