|
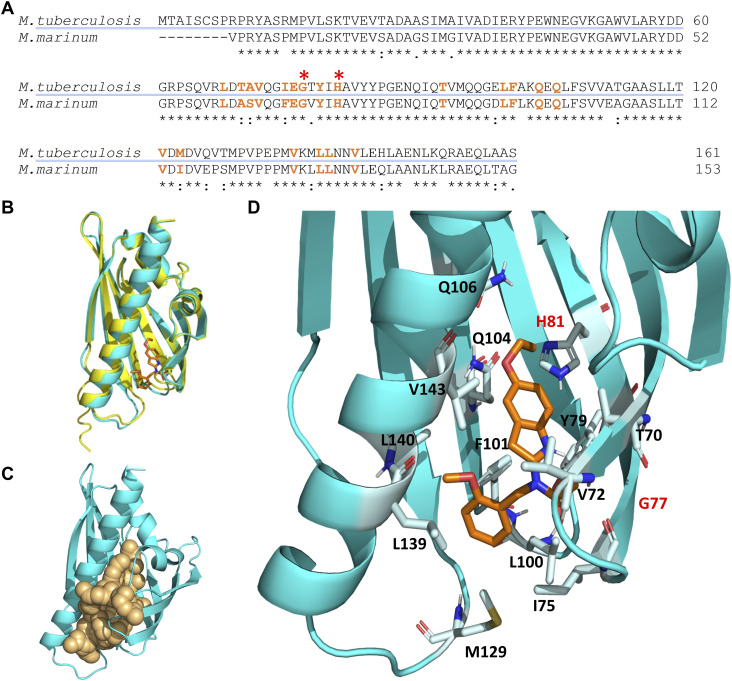
Figure 5. Molecular docking simulation of the structure of the Rv0164 complex with BT-37.

|
|
Figure 5. Molecular docking simulation of the structure of the Rv0164 complex with BT-37.