
- Title
-
uhrf1 and dnmt1 Loss Induces an Immune Response in Zebrafish Livers Due to Viral Mimicry by Transposable Elements
- Authors
- Magnani, E., Macchi, F., Madakashira, B.P., Zhang, C., Alaydaroos, F., Sadler, K.C.
- Source
- Full text @ Front Immunol

|

|
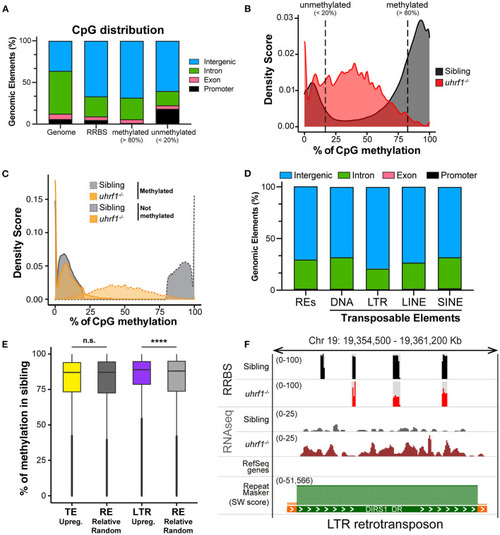
DNA methylation is enriched on TEs that become activated in |

|

Viral signaling pathways are required for the gene expression and cell death phenotypes in |