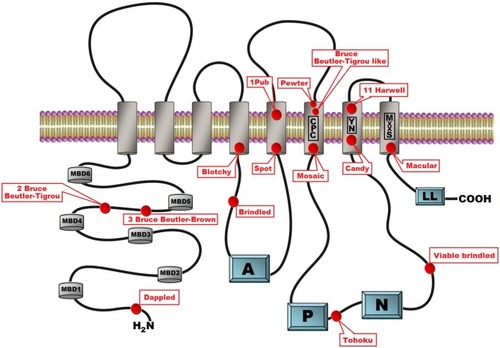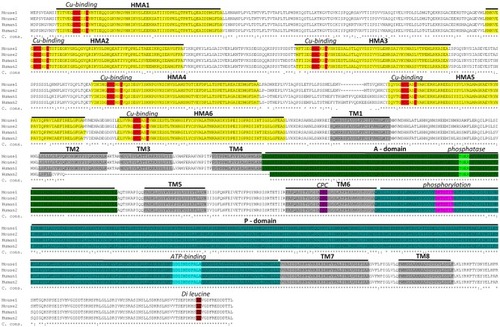
Sequence alignment between mouse and human ATP7A proteins. Two murine isoforms (NP_001103227.1 (Mouse 1) and NP_033856.3 (Mouse 2) and two human isoforms (NP_000043.4 (Human 1) and NP_001269153.1 (Human 2) are shown. Functional domains: HMA—heavy-metal-associated domain, TM—transmembrane domain, A-domain—actuator domain, P-domain—phosphorylation domain and corresponding functional motifs: Cu-binding, phosphatase, phosphorylation, ATP-binding and di-leucine are marked. It should be noted, that the human isoform 2 is shorter than isoform 1 and lacks a part of TM2 in addition to TM3 and TM4. This product is a result of alternative splicing, leading to skipping of exon 10. Isoform 2 is expressed at low level, in normal healthy individuals, but has been observed as the major product in a patient with a IVS10 mutation. Because the patient had OHS, in contrast to classic Menkes Disease, it has been suggested that isoform 2 has partly copper transporting activity (Qi and Byers, 1998). The sequence alignment is performed using Clustal Omega software (http://www.ebi.ac.uk/Tools/msa/clustalo/). Characteristic protein domains are marked based on conserved domains database (Marchler-Bauer et al., 2015) and previously published ATP7A protein structures (Kaler, 2011; Tümer, 2013). C. cons. – Clustal Omega consensus. An “*” (asterisk) indicates positions with fully conserved residues, a “:” (colon) indicates conservation between groups of strongly similar properties and a (period) indicates conservation between groups of weakly similar properties.
|