
- Title
-
Disrupting the Repeat Domain of Premelanosome Protein (PMEL) Produces Dysamyloidosis and Dystrophic Ocular Pigment Reflective of Pigmentary Glaucoma
- Authors
- Hodges, E.D., Chrystal, P.W., Footz, T., Doucette, L.P., Noel, N.C.L., Li, Z., Walter, M.A., Allison, W.T.
- Source
- Full text @ Int. J. Mol. Sci.

ZFIN is incorporating published figure images and captions as part of an ongoing project. Figures from some publications have not yet been curated, or are not available for display because of copyright restrictions. EXPRESSION / LABELING:
PHENOTYPE:
|
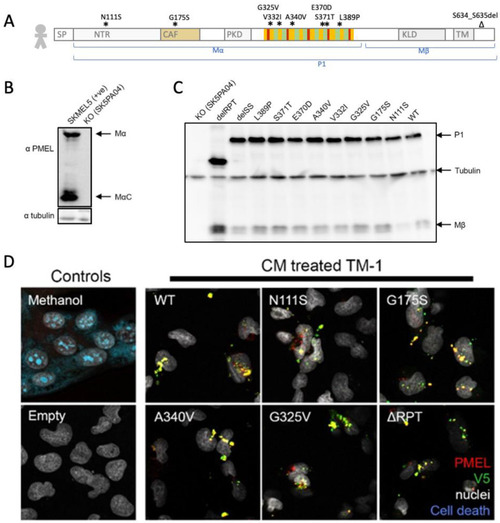
Human PMEL is engulfed by trabecular meshwork cells, providing a |
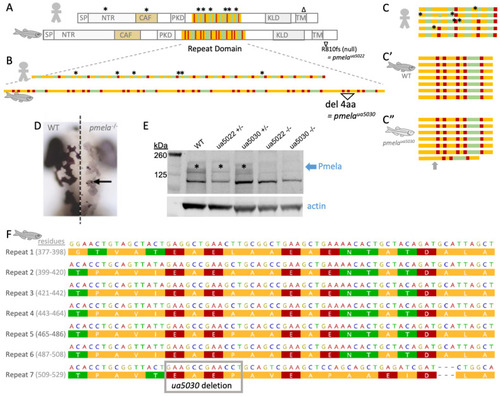
A zebrafish model of pigmentary glaucoma engineered via subtle mutation to the repeat region of the EXPRESSION / LABELING:
PHENOTYPE:
|
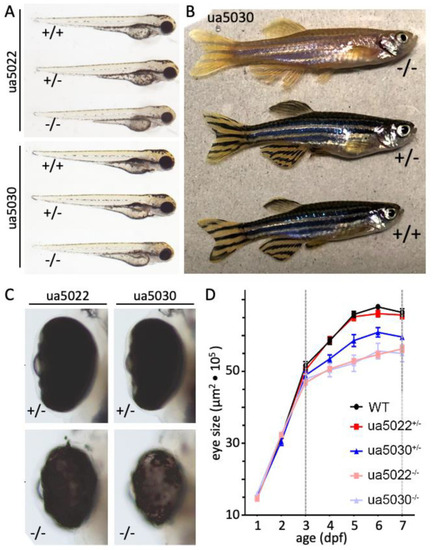
PHENOTYPE:
|
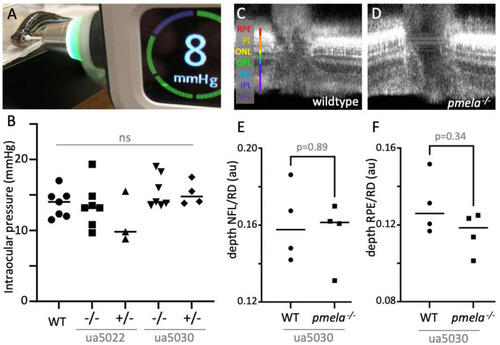
Adult PHENOTYPE:
|
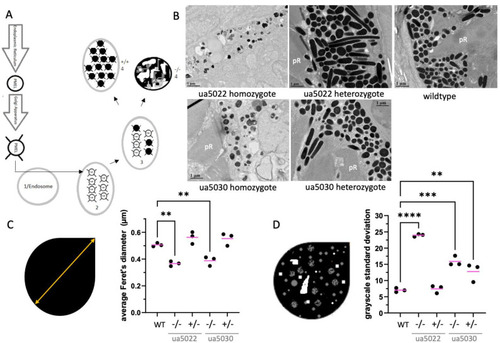
The repeat region of the Pmela protein is required for the elongation of melanosomes and the even distribution of melanin within melanosomes. ( PHENOTYPE:
|
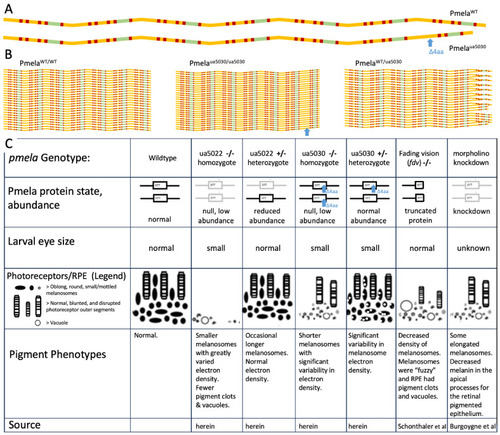
Summary of |