
Figure 1:
- ID
- ZDB-FIG-211129-25
- Publication
- Schnur et al., 2021 - UBA2 variants underlie a recognizable syndrome with variable aplasia cutis congenita and ectrodactyly
- Other Figures
- All Figure Page
- Back to All Figure Page
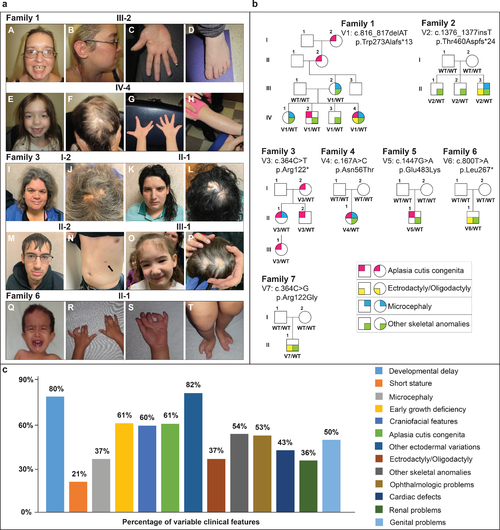
Fig. 1. Clinical phenotypes and pedigrees associated with UBA2-related syndrome. (a) Family 1, III-2: A. prominent forehead, high hairline, discolored peg-shaped teeth with gap between upper incisors, cleft chin. B. Low-set ear with simple cartilaginous pattern. C. diminished distal flexion creases. D. Brachydactyly, mild 2–3 syndactyly, clinodactyly of the 4th toe. Family 1, IV-4: E. Prominent forehead, high hairline, cleft chin, mildly downslanted palpebral fissures. F. Aplasia cutis congenita (ACC). G. Repaired ectrodactyly, hypoplastic distal flexion creases. H. Ichthyosis. Family 3, I-1: I. tall forehead, hypertelorism, broad nasal root, mild micrognathia. J. ACC. Family 3, II-1: K. Tall forehead, hypertelorism, broad nasal root, thin upper lip, medial eyebrow flare. L. ACC. Family 3, II-2: M. Facial features. N. Supernumerary nipple (arrow). Family 3, III-1: O. Tall forehead, low-set ears, micrognathia. P. ACC. Family 6, II-1: Q. High forehead, hypertelorism, bilateral epicanthal folds. R, S. bilateral 2–3 finger syndactyly, camptodactyly T. bilateral ectrodactyly of the feet. (b) Affected individuals are shown as filled symbols. Genotypes are shown below each individual who was genotyped. (c) Percentages of different clinical features variably expressed in UBA2-affected individuals based on available data. Previously reported UBA2 patients are also included in the percentages. |