
|
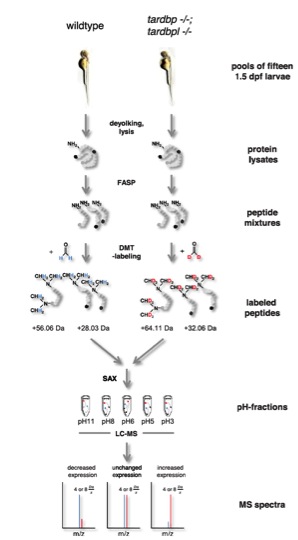
Fig. S7 Schematic view of the quantitative proteomic analysis. Pools of 16 1.5-dpf embryos were deyolked and lysed consecutively. Protein lysates were tryptically digested using the filter-assisted sample preparation technique (FASP). Tryptic peptides ending either on arginine (depicted in black) or a lysine (depicted in dark gray) were labeled using differential stable isotope dimethyl labeling (DMT-labeling). Peptides gained from wild-type embryos were labeled “light” using undeuterated formaldehyde, whereas peptides gained from tardbp-/-;tardbpl-/- mutant embryos were labeled “heavy” using deuterated formaldehyde. Dimethylation occurs at free amine-groups resulting in the addition of four methyl-groups to lysine-containing tryptic peptides whereas only two methyl-groups are added to arginine-containing tryptic peptides; this results in a shift of +56.06 Da or +28.03 Da, respectively, for the light-labeledpeptides. Heavy-labeled peptides are shifted by further +8 Da or +4 Da, respectively, because of the two deuterium atoms contained in every methyl-group. After labeling, peptides from both experimental groups were combined and fractionated using stage-tip-based anion-exchange chromatography (SAX). Mass spectrometry was done for every of the five pH-fractions in two technical replicates. Abundance levels of peptides can be calculated from the intensity ratio between the isotope clusters resulting from the light and heavy peptides.