
- Title
-
Loss-of-function mutations in UDP-Glucose 6-Dehydrogenase cause recessive developmental epileptic encephalopathy
- Authors
- Hengel, H., Bosso-Lefèvre, C., Grady, G., Szenker-Ravi, E., Li, H., Pierce, S., Lebigot, É., Tan, T.T., Eio, M.Y., Narayanan, G., Utami, K.H., Yau, M., Handal, N., Deigendesch, W., Keimer, R., Marzouqa, H.M., Gunay-Aygun, M., Muriello, M.J., Verhelst, H., Weckhuysen, S., Mahida, S., Naidu, S., Thomas, T.G., Lim, J.Y., Tan, E.S., Haye, D., Willemsen, M.A.A.P., Oegema, R., Mitchell, W.G., Pierson, T.M., Andrews, M.V., Willing, M.C., Rodan, L.H., Barakat, T.S., van Slegtenhorst, M., Gavrilova, R.H., Martinelli, D., Gilboa, T., Tamim, A.M., Hashem, M.O., AlSayed, M.D., Abdulrahim, M.M., Al-Owain, M., Awaji, A., Mahmoud, A.A.H., Faqeih, E.A., Asmari, A.A., Algain, S.M., Jad, L.A., Aldhalaan, H.M., Helbig, I., Koolen, D.A., Riess, A., Kraegeloh-Mann, I., Bauer, P., Gulsuner, S., Stamberger, H., Ng, A.Y.J., Tang, S., Tohari, S., Keren, B., Schultz-Rogers, L.E., Klee, E.W., Barresi, S., Tartaglia, M., Mor-Shaked, H., Maddirevula, S., Begtrup, A., Telegrafi, A., Pfundt, R., Schüle, R., Ciruna, B., Bonnard, C., Pouladi, M.A., Stewart, J.C., Claridge-Chang, A., Lefeber, D.J., Alkuraya, F.S., Mathuru, A.S., Venkatesh, B., Barycki, J.J., Simpson, M.A., Jamuar, S.S., Schöls, L., Reversade, B.
- Source
- Full text @ Nat. Commun.

|
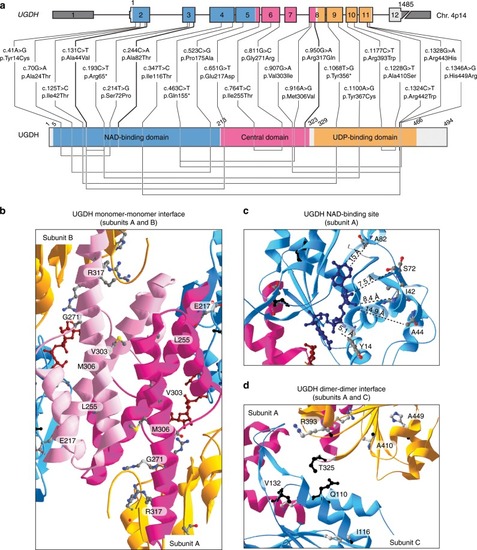
|
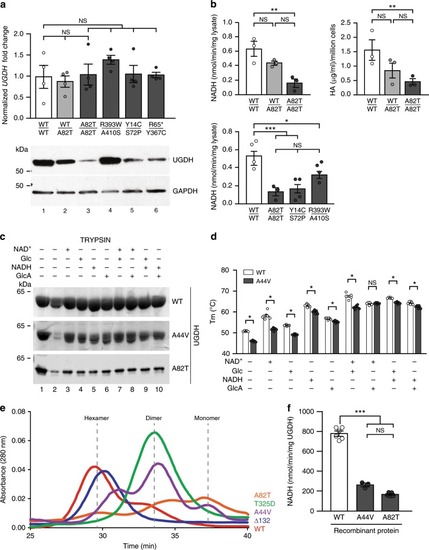
|

|

ZFIN is incorporating published figure images and captions as part of an ongoing project. Figures from some publications have not yet been curated, or are not available for display because of copyright restrictions. EXPRESSION / LABELING:
PHENOTYPE:
|