
Fig. 1
- ID
- ZDB-IMAGE-201012-108
- Publication
- Pini et al., 2020 - ALX1-related frontonasal dysplasia results from defective neural crest cell development and migration
- All Figures
- Figures for Pini et al., 2020
|
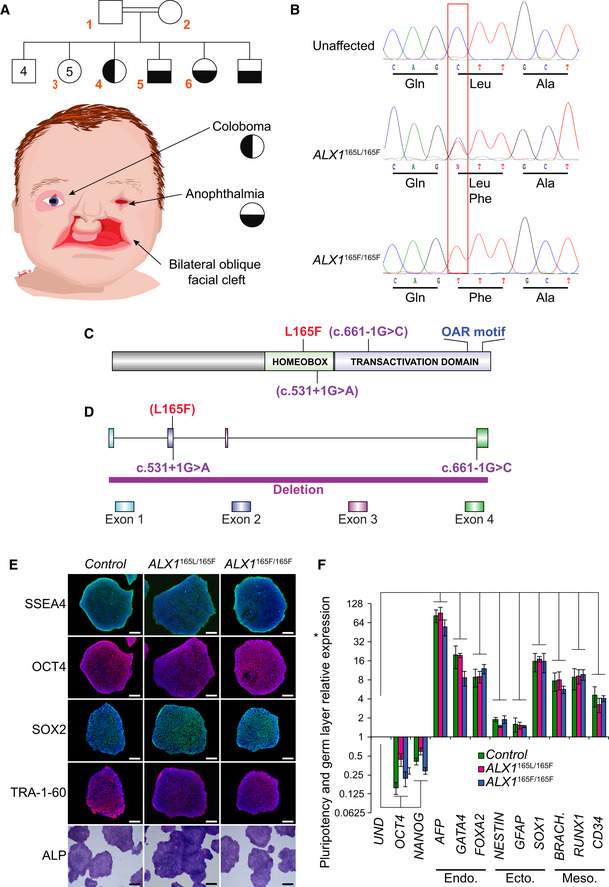
Fig. 1
The pedigree family tree includes two unaffected parents, four unaffected male siblings, five unaffected female siblings, and two each female and male affected sibling. Subjects 1–6, indicated in red, were enrolled in the study. Subjects 4–6 show complex FND with ocular involvement. The eldest affected sibling (subject 4) presented with right coloboma, left microphthalmia, and bilateral Tessier 4 oblique facial clefts. Subject 5 presented with bilateral anophthalmia with fused eyelids and shallow orbits, with bilateral oblique facial clefts. Subject 6 presented with bilateral anophthalmia with open shallow orbits, absent upper and lower eyelids, exposed orbital mucosa, bilateral oblique facial clefts, and malformed nasal ala with nodular skin tags. iPSCs were generated using blood samples collected from subjects 1, 5, and 6. Whole‐exome sequencing was carried out and analysis revealed a missense p.L165F variant (c.493 C>T) in the Schematic of the ALX1 protein structure showing the position of the L165F substitution described here (red) and the locations of exon borders affected by two reported pathogenic variants (purple; Ullah Schematic of the Immunofluorescence staining for pluripotent markers Expression of pluripotent (OCT4, NANOG), endoderm (Endo., AFP, GATA4, FOXA2), ectoderm (Ecto., NESTIN, GFAP, SOX1), and mesoderm (Meso., BRACH. (BRACHYURY), RUNX1, CD34) gene markers for