|
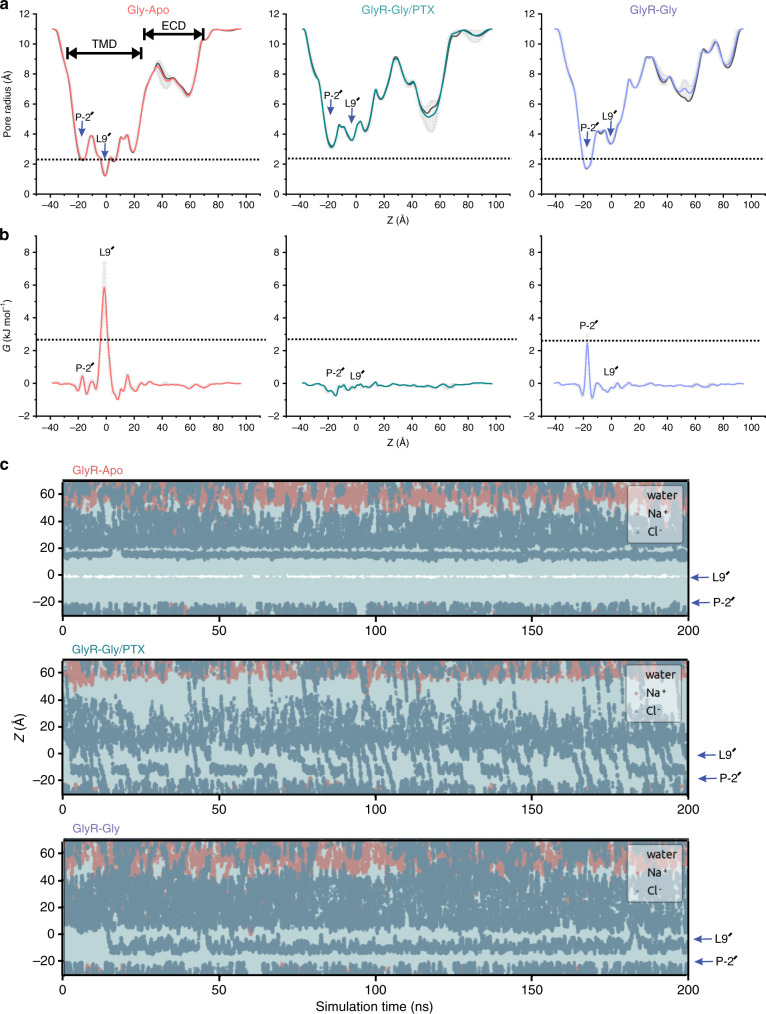
Fig. 6

|
|
Fig. 6