
- Title
-
Animal Models of Ehlers-Danlos Syndromes: Phenotype, Pathogenesis, and Translational Potential
- Authors
- Vroman, R., Malfait, A.M., Miller, R.E., Malfait, F., Syx, D.
- Source
- Full text @ Front Genet
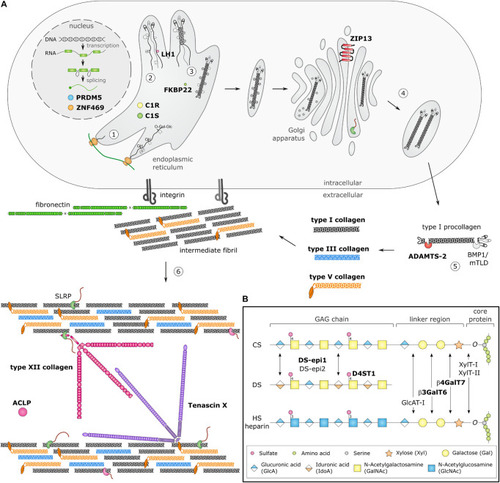
Schematic overview of collagen and glycosaminoglycan (GAG) biosynthesis and collagen fibrillogenesis. Molecules defective in Ehlers-Danlos syndromes (EDS) are highlighted in bold. |
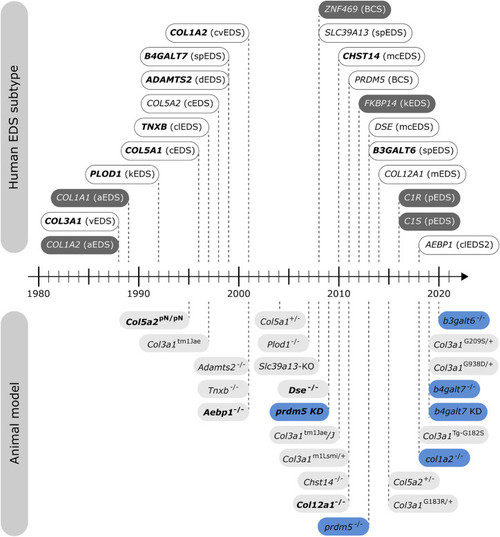
Timeline illustrating the first identification of molecular defects in human EDS (above timeline) and the first description of engineered animal models targeting an EDS-associated gene (below timeline). Human genes associated with EDS without a corresponding animal model are indicated in dark gray. Note that for some human EDS subtypes, biochemical and/or ultrastructural findings preceded the identification of the molecular defect. Mouse models are depicted in light gray and zebrafish models in blue. Bold indicates what was first, either the identification of the human disease gene or the generation of the engineered animal model. |
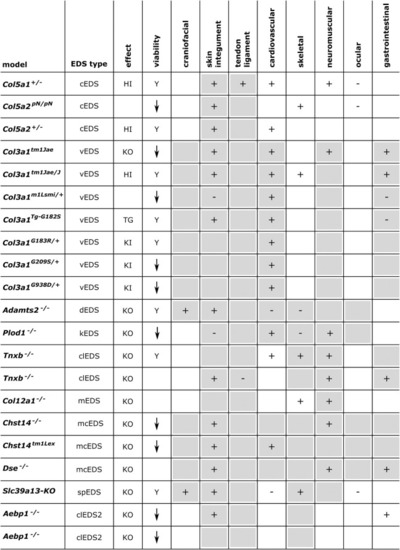
|
Overview of the phenotypic findings in some mouse models of EDS. |
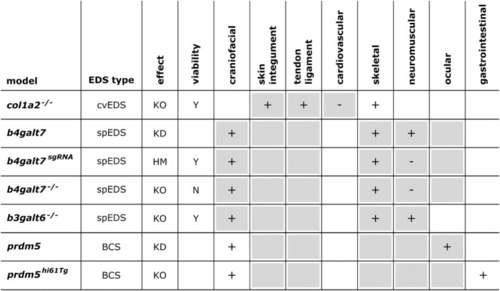
Phenotypic characteristics of zebrafish models with defects in EDS-associated genes. The presence or absence of a phenotype in the zebrafish model is indicated with “+” or “-”, respectively, when investigated. A detailed description of the zebrafish phenotypes can be found in |

Overview of the phenotypic findings in zebrafish models of EDS. |