
- Title
-
Zebrafish mutants calamity and catastrophe define critical pathways of gene-nutrient interactions in developmental copper metabolism
- Authors
- Madsen, E.C., and Gitlin, J.D.
- Source
- Full text @ PLoS Genet.

Chemical genetic screen for zebrafish mutants sensitive to copper deficiency. (A) Diagram outline of the sensitivity screen. Haploid embryos are either wild-type (green) or mutant (yellow) for any given ENU-induced mutation. These embryos were placed in vehicle or 100 nM neocuproine (neoc) at 3 hpf and allowed to develop until 48 hpf when they were screened for loss of melanin pigmentation in drug only. The ideal mutant is demonstrated on the right where mutant (yellow) embryos have no pigment only upon treatment with neocuproine (B–C) The first mutant isolated has full pigmentation without neocuproine (B) and loses all pigmentation upon treatment with 100 nM neocuproine (C). (D) This mutant does not complement a known allele of the mutant calamity, establishing it as a new allele of the same gene atp7a. (E–F) The second mutant has reduced, punctate pigmentation without drug treatment (E) but loses all pigmentation upon treatment with 100 nM neocuproine (F). (G) The second mutant fully complements calamity. Therefore we have isolated a new mutant which we have called catastrophe. PHENOTYPE:
|
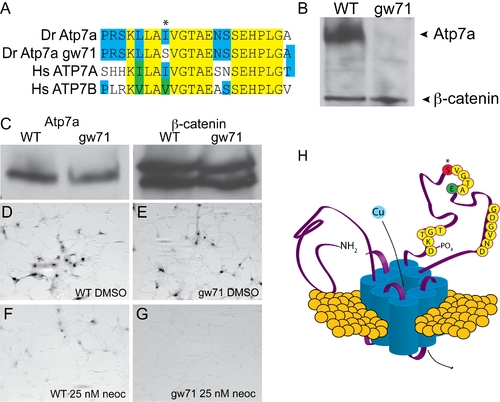
calamitygw71 contains a hypomorphic allele of atp7a. (A) The mutation in calgw71 is a T1061S substitution in a highly conserved region of the vertebrate copper transporters and exchanges a normally hydrophobic amino acid for a hydrophilic one (asterisk). (B) This single amino acid change results in near complete loss of immunoblot-detectable protein levels. Wild-type and gw71 mutant embryos were blotted for Atp7a using a peptide antibody to the C-terminus of the protein. β-catenin was used as a loading control. (C) Despite loss of protein in the zebrafish, transfection of an ATP7A-deficient human fibroblast cell line with either wild-type or mutant cDNA (derived from site-directed mutagensis of the wild-type) results in near equivalent expression of the zebrafish protein. β-catenin is again used as a loading control. (D–E) Both wild-type (D) and mutant (E) cDNAs are capable of producing functional protein as measured by functional tyrosinase activity in ATP7A deficient fibroblasts fixed and stained with L-DOPA. (F–G) Both wild-type (F) and mutant (G) Atp7a are sensitive to the effects of low-dose neocuproine in the above assay; however, the mutant cDNA is much more sensitive to mild copper chelation (F vs. G). (H) Model illustrating the relationship of the mutation (red, asterisk) to the known topology and functional domains of Atp7a. The mutation lies in the ATPase domain of the protein near a glutamate (green “E”) required for ATP binding and hydrolysis [10]. EXPRESSION / LABELING:
|

calamitygw71 embryos display developmental defects that are sensitive to maternal and environmental copper availability. (A–D) Maternal effect on pigmentation in untreated gw71 homozygous embryos. Mutant embryos derived from a heterozygous mother (A) display near normal pigmentation and have an incomplete loss of pigmentation in 100 nM neocuproine (B) most noticeable in the retina (arrowhead). Mutant embryos derived from homozygous mothers have mild hypopigmentation (C), particularly of the retina (arrowhead) and lose nearly all pigmentation in 100 nM neocuproine (D). (E–I) Partially penetrant juvenile skeletal deformities are present in gw71 mutant fish. Wild-type (E) and gw71 mutant (F) 21 dpf larvae were stained with alcian blue (cartilage) and alizarin red (bone) to reveal skeletal defects. In (G), embryos were untreated, treated with 100 nM neocuproine, or treated with 500 nM CuCl2 during the times indicated. The larvae at 21 dpf were scored according to absence or presence of a vertebral axis defect. A one-tailed Fisher exact probability test was used to calculate p-values. Only the indicated p-value was significant. (H) gw71 mutants at an earlier stage of bone ossification display hyperossification at the location of the vertebral defect (arrow). Normal ossification is detected by alizarin red staining and begins rostrally (arrowhead). (I) A higher magification of the defect in (H) showing the hyperossification (arrow) and an outpouching of connective tissue which stains with alcian blue (arrowhead). PHENOTYPE:
|
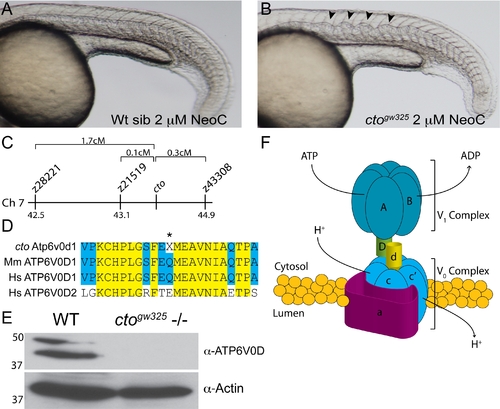
catastrophegw325 contains a copper sensitive mutation in the vacuolar (H+) ATPase Atp6. (A–B) cto embryos are globally sensitive to copper deficiency. Wild-type embryos (A) in 2 μM neocuproine do not display notochord defects. In contrast, cto embryos (B) placed in this same dose of neocuproine have significant distortion of the notochord in a pattern consistent with loss of lysyl oxidase activity [12]. (C) Meotic mapping placed the cto mutation between markers z21519 (43.1cM) and z43308 (44.9cM) on chromosome 7. (D) Atp6v0d1 is highly conserved between zebrafish and mammals and is easily differentiated from ATP6V0D2 present in humans. The amino acid Q136 is changed to a stop in the mutant (asterisk). (E) The mutation in cto abolishes expression of the full length protein. Immunoblot analysis using a C-terminal polyclonal antibody shows no recognition of the 40 kD band in 48 hpf cto embryos. The identity of the band at 50 kD is unknown. Actin was used as a loading control (lower panel). (F) Model of the proposed quaternary structure of Atp6. The lower-case d subunit (yellow) forms part of a connecting stalk between the V1 and V0 subunits the presence of which is required for proper formation of the entire transporter [19]. |
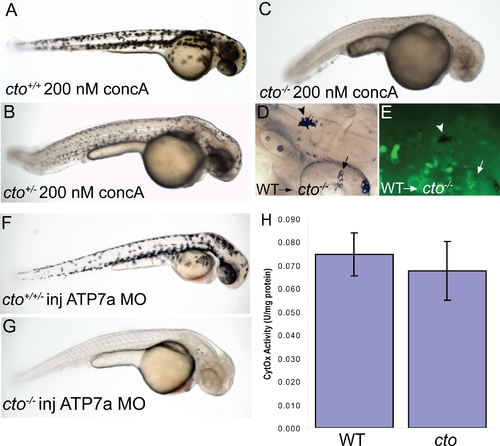
cto contains a concanamycin A sensitive, cell autonomous defect which affects secretory pathway copper transport. (A–C) cto gene dosage alters sensitivity to concanamycin A (concA) an inhibitor of Atp6. Wild-type fish have no phenotypic response when incubated in 200 nM concanamycin A beginning at 24 hpf (A). Embryos heterozygous for cto are sensitive to this same dose of concA, resulting in punctate melanocytes (B). ConcA exacerbates the phenotype of catastrophe homozygotes resulting in total loss of pigmentation and increased degenerative appearance (C). (D–E) The defect in cto is cell autonomous both in epidermal and retinal pigment epithelial cells. Wild-type, GFP-positive cells were transplanted into cto mutant embryos at the 1000 cell stage and allowed to develop to 48 hpf. Robustly pigmented melanocytes with normal size and shape can be seen sparsely distributed throughout the epidermis (D, arrowhead) and retinal pigment epithelium (arrow, D). The epidermal melanocyte does not have visible GFP but is not surrounded by GFP-positive cells (E, arrowhead). The RPE cells have a central area of GFP-positivity (E, arrow) Other areas are GFP positive without melanin pigment. (F–G) cto homozygotes but not heterozygotes or wild-type embryos are sensitive to atp7a morpholino injection. At a sensitizing dose of morpholino that does not affect wild-type/heterozygotes (F), homozygous cto embryos lose all pigmentation (G). (H) Cytochrome c oxidase activity is not reduced in cto embryos. Activity was normalized to protein levels in each sample. Three independent samples were prepared from three groups of embryos and the standard deviation of the three experiments is shown. PHENOTYPE:
|
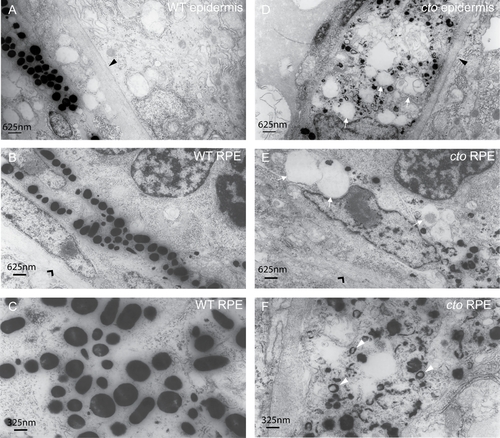
cto melanocytes have significant ultrastructural defects. (A–B) Wild-type epidermal (A) and retinal pigment epithelial (RPE, B) melanocytes are elongated and thin and contain many large, densely pigmented melanosomes. The epithelial basement membrane is indicated by a black triangle in (A) and the RPE basement membrane by a black arrowhead in (B). (C) A higher magnification of wild-type melanosomes showing significant pigmentation and ellipsoid shape when cut longitudinally. (D–E) cto mutant epidermal (D) and RPE (E) melanocytes showing rounded, poorly pigmented cells that contain numerous large, empty vesicles (white arrows) The basement membranes are indicated as in (A and B). (F) A higher magnification of RPE melanosomes showing the diverse array of immature, poorly pigmented vesicles. The white arrowheads point to multi-lamellar, melanin filled vesicles which are identical to the melanin positive multi-vesicular bodies seen in the cappucino mouse (see text). PHENOTYPE:
|
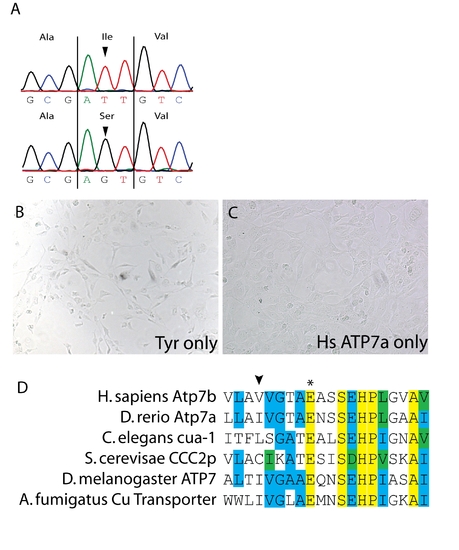
(A) Sequencing of the atp7a cDNA in calgw71 mutant embryos reveals a single non-synonymous nucleotide change T3182G which causes a non-conservative amino acid substitution T1061S. (B) Transfection of tyrosinase only into Me344 cells does not result in any appreciable tyrosinase activity. (C) Transfection of atp7a only into Me344 cells also does not result in L-DOPA oxidase activity. This activity is specific to tyrosinase expression. (D) Alignment of a small region of atp7a containing the mutation in gw71 (arrowhead) and the highly conserved glutamate (asterisk) observed to be important for ATP binding/hydrolysis. This glutamate is fully conserved from fungus to humans. |

Unillustrated author statements PHENOTYPE:
|